Comprehensive Review of Contrastive and Generative Self-Supervised Learning for Small Molecular Representation
https://t.co/Jwov2TIIG2
#JCIM Vol66 Issue11 #Review
🧑💻🧑🏫 I'm recruiting a post-doc to work on Tabular Foundation Models, one of the hotest topics in AI, where we are at the leading edge.
This is an opportunity to develop the next-level tabular AI, blending deep learning and tables.
Assessment of Alphafold Protein Models for Small-Molecule Ligand Docking versus Co-Folding
https://t.co/JgztzLartq
#JCIM Vol66 Issue10 #MachineLearning#DeepLearning
Together with UC Berkeley we are announcing the laser phase plate - a breakthrough in atomic resolution imaging. This is the brightest continuous wave laser in the world, 100 million times the intensity of the surface of the sun.
Phase contrast plays an important role in microscopy, but it was thought close to impossible for electron microscopy, where it would require interfering with an electron beam. Holger Mueller and Robert Glaeser proposed exactly this using a standing wave laser. It has taken over 15 years to make this a reality. Biohub partnered with UC Berkeley and Mueller to support this work and to engineer and build the technology.
Contrast has been the critical barrier to achieving atomic resolution imaging of the cell. In cryo-electron tomography, a cellular imaging technology that uses electron microscopy, the low contrast makes it impossible to resolve anything but the largest proteins within their cellular context. The laser phase plate removes that barrier.
With advances in AI this breakthrough in contrast will start to open up a new frontier in structural biology, that will allow us to see the molecular machines of the cell, and how they assemble into far more complex and dynamic systems, and understand how they work.
AMD Ryzen AI Halo. The ultimate local AI developer platform.
Pre-order now: https://t.co/Ny0ZV8LOYi
⚡ Up to 128GB unified memory
⚡ Support for models up to 200B parameters
⚡ Windows & Linux support
⚡ Ready-to-run AI workflows out of the box
Build, prototype, and deploy locally without cloud constraints.
110 million DFT calculations: the dataset that cured a stubborn bias in materials ML
Predicting whether a new material is stable, or how it conducts heat, usually means running density functional theory (DFT): accurate quantum mechanics, but painfully slow. Machine learning interatomic potentials (MLIPs) promise to act as fast surrogates for DFT, yet they share a stubborn weakness. Most are trained on databases of relaxed, near-equilibrium structures, so when a real simulation pushes them toward distorted, far-from-equilibrium configurations, they degrade. The community even named the symptom: "systematic softening," a consistent underprediction of energies, forces, and vibrational frequencies.
Luis Barroso-Luque and coauthors attack the problem from the data side rather than the model side. They release OMat24, an open dataset of over 110 million DFT calculations spanning most of the periodic table, built deliberately around non-equilibrium structures generated through rattled Boltzmann sampling, short ab initio molecular dynamics, and re-relaxations of perturbed crystals. The whole design bet is on diversity: structures far from the comfortable energy minima where older datasets cluster.
The payoff is concrete. MLIPs pretrained on OMat24 top the Matbench-Discovery leaderboard, with F1 scores above 0.9 for stability and formation-energy errors near 18 to 20 meV/atom. More telling is that this data diversity largely eliminates systematic softening across very different model architectures, and models trained on OMat24 alone soften even less than fine-tuned variants. Within months of release, every leading model on the leaderboard had adopted the dataset.
For materials development, batteries, energy, or catalysis, the takeaway is that the limiting factor for trustworthy property prediction is often the training data, not a cleverer network. Building or reusing diverse, far-from-equilibrium data as a pretraining foundation can make screening pipelines reliable under the operating conditions that actually matter, instead of only at idealized minima.
Paper: Barroso-Luque et al., Nature Computational Science (2026) — journal license | https://t.co/6vgGBqye6S
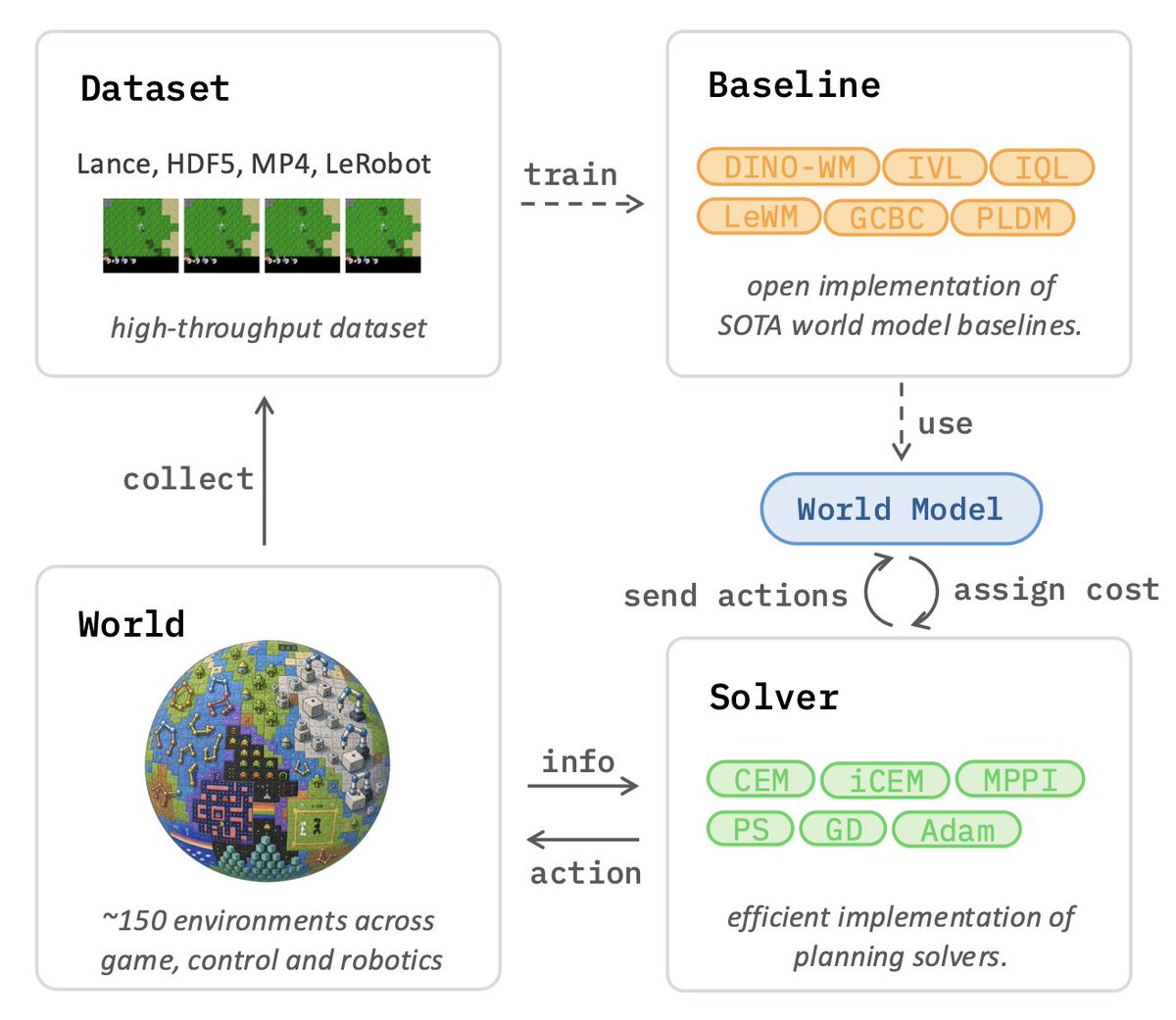
Would you like to join the research effort on JEPA and World Models easily?
After a full year of hard work, we’re excited to finally release stable-worldmodel:
an open-source, scalable platform built to accelerate JEPA & World Model research!
📄: https://t.co/gnxGvens5A
Divide and Cluster: The DIVINE Framework for Deterministic Top-Down Analysis of Molecular Dynamics Trajectories #MolecularDynamics
https://t.co/c6ItNZVnZE
@GroupQuintana@UF#JCIM Vol66 Issue8 #compchem
A dataset of 1.2 million molecules with DFT-level quantum chemical annotations for molecular representation learning
1 qcMol is introduced as a 1,200,216-molecule dataset designed for molecular representation learning, aiming to close the gap between quantum-chemistry datasets (often small or chemically artificial) and real-world drug discovery needs (often lacking physics-grounded electronic descriptors).
2 The key differentiator is multi-scale quantum annotation: 31 DFT-derived properties per molecule, split into 15 global descriptors (e.g., HOMO-LUMO gap, dipole moment, shape/isosurface metrics) and 16 local descriptors (atom- and bond-level) that can be directly used in graph-based ML.
3 Local electronic structure is made ML-friendly via wavefunction post-analysis: NBO and Multiwfn extract structured, chemically interpretable node/edge features such as NAO-related descriptors, localization index (LI), delocalization index (DI), electron localization function (ELF), Laplacian bond order (LBO), and related bond/orbital terms.
4 The dataset emphasizes chemical/biological relevance rather than purely enumerated chemical space: it aggregates molecules from 95 public datasets spanning molecular properties, intermolecular interactions, molecule-cell interactions, drug-like compounds, and metabolites (including HMDB and PAMDB), with 63.65% of molecules linked to systematic experimental results.
5 Diversity is a central design goal: qcMol reports 247,448 unique Bemis–Murcko scaffold types (avg 4.85 molecules/scaffold) and covers a broad size range (1 to 604 atoms), extending far beyond common quantum datasets constrained to very small organics; elements are restricted to main-group up to the 5th period, with ECPs used where appropriate.
6 Quantum chemistry pipeline balances scale and fidelity: RDKit/Open Babel generate initial 3D structures, GFN2-xTB performs geometry optimization (extreme settings), then ORCA runs single-point DFT at B3LYP-D3/def2-SV(P), followed by NBO 7.0 + Multiwfn 3.8 wavefunction analysis to compute global/local descriptors.
7 Benchmarking focuses on practical ADMET prediction: using GAT across 6 MoleculeNet tasks (FreeSolv, ESOL, Lipophilicity, ClinTox, BBBP, BACE), adding qcMol QC features to standard RDKit descriptors improves performance across most configurations and also speeds up training convergence; ablations suggest atom-level QC features contribute most, with NBO-related edge features particularly important among bond descriptors.
8 Distribution-level evaluation targets pretraining realism: SchNet encoders pre-trained on 50k molecules from qcMol vs QM9 (masked reconstruction) transfer better to downstream ADMET tasks in most settings, with clearer advantages on larger molecules (>40 atoms), supporting the claim that qcMol better matches real-world chemical space for representation learning.
9 The resource is delivered as a database with browsing/search/download plus an upload interface to request new annotations; data are organized per molecule into basic info, global features, and local graph-structured features, with optional access to calculation artifacts (inputs/outputs and wavefunction-related files) for deeper analysis and reproducibility.
💻Code: https://t.co/tQORZQ9oBt
📜Paper: https://t.co/bqHl97xyLx
#ComputationalChemistry #QuantumChemistry #DFT #MolecularRepresentation #GraphNeuralNetworks #Cheminformatics #DrugDiscovery #ADMET #MachineLearning #Datasets
For over a decade, we’ve accepted that end-to-end backprop is the only way to train deep networks. But holding the entire network in memory all at once is why AI training is hitting a resource wall.
We found a new way to break the network into blocks and train them independently. The trick? Treating the network’s forward pass like a diffusion model denoising a signal.
This reinterpretation slashes the memory needed to train deep models. In our #ICLR2026 paper (https://t.co/PK5h0mqQSo), we matched end-to-end performance across ViTs, DiTs, and LLMs. We did this while training just one isolated block at a time.
Connect Claude to PDBe in 3 steps:
1️⃣ Install uv
2️⃣ Run:
uvx pdbe-mcp-server --server-type pdbe_api_server
3️⃣ Add config to Claude Desktop
Now ask:
“Find X-ray structures for UniProt P12345”
📦 https://t.co/NcwR2h1ipu
#PDBe#MCP#ClaudeAI#Bioinformatics
AI agents are advancing research-level math. 🚀
I’m thrilled to share @GoogleDeepMind’s AlphaProof Nexus - an agentic framework for formal proof search powered by Gemini.
When applied to a set of open formal math problems, our agent autonomously solved:
✅ 9 open Erdős problems (including two open for 56 years!)
✅ 44 Online Encyclopedia of Integer Sequences (OEIS) problems
✅ A 15-year-old open problem in algebraic geometry ✅ A 7-year-old open question in min-max optimization
We are collaborating with mathematicians across disciplines - from combinatorics and graph theory to quantum optics. Ultimately, these results show the massive potential of even simple agentic loops powered by Gemini.
Read the paper here: https://t.co/c5M9ZjRXU1
SMDD-Bench: Can LLMs Solve Real-World Small Molecule Drug Design Tasks?
1. The paper introduces SMDD-Bench, a multi-turn, long-horizon benchmark designed to test whether LLM agents can handle realistic small-molecule drug design workflows, rather than single-turn chemistry QA. It contains 502 guaranteed-solvable task instances spanning diverse chemistries and 102 protein targets.
2. The most striking result: even the best tested model (GPT-5.4) solves only 40.2% overall, despite using a tool-enabled agent scaffold. Performance concentrates heavily in Lead Optimization, while 3D-heavy tasks (Interaction Point Discovery, Scaffold Hopping, Fragment Assembly) remain near-zero for most models.
3. SMDD-Bench covers five task types that map to core medicinal chemistry loops: 2D Pharmacophore Identification, Interaction Point Discovery (3D pocket hotspots), Scaffold Hopping (new scaffold, same binding mode), Lead Optimization (multi-property optimization under constraints), and Fragment Assembly (link/expand fragments while preserving poses).
4. A key methodological contribution is witness-aware task generation: for task types where solvability is not guaranteed by default (Scaffold Hopping, Lead Optimization, Fragment Assembly), each instance is constructed alongside a hidden “witness” molecule that is known to pass the same evaluation pipeline. This makes every task instance solvable by construction, enabling scale without manual curation.
5. Evaluation is fully automated and tool-grounded: RDKit and OpenBabel for chemistry checks, PLIP for interaction fingerprints, Boltz2 for co-folded protein–ligand structures plus binding probability/affinity, and ADMET-AI for 8 ADMET endpoints (e.g., hERG, CYP3A4, solubility, clearance, BBB, Ames). Agents operate under limited oracle budgets to mimic scarce wet-lab feedback.
6. Lead Optimization is formulated as constrained multi-objective optimization: improve up to 5 properties, hold up to 4 within tolerance bands, satisfy hard drug-likeness constraints (MW, logP, TPSA, HBD/HBA, rotatable bonds, charge, SA score, PAINS/Brenk/NIH alerts), and remain in-series via a Tanimoto similarity constraint to the reference (≥0.7).
7. The benchmark also surfaces a practical agent failure mode: enumeration vs. selection. By retrospectively scoring all SMILES an agent mentioned (including those it did not submit to oracles), the authors show “recovered” success rates can be much higher—especially for Scaffold Hopping—implying agents often generate viable candidates but fail to choose them without extra oracle calls.
8. Output novelty is quantified against major public chemistry databases (ChEMBL, SureChEMBL, PubChem, BindingDB). Many models’ successful submissions are largely novel by this conservative check, but novelty alone is not sufficient: agents still struggle to satisfy 3D interaction constraints and multi-turn planning under budget.
9. Diversity is evaluated via SMDD-Bench Diversity (20 hard Lead Optimization tasks, 10 runs each). Models frequently converge to the same solutions; the benchmark reports unique-success counts and pairwise similarity, highlighting that “many parallel agents” may not explore chemical space as broadly as desired in real campaigns.
10. The authors provide SMDD-Bench Lite (100 instances) to reduce compute barriers, and document common failure modes beyond raw accuracy: lack of cross-turn SAR synthesis (re-testing disqualified motifs), incoherent multi-turn planning (not building on previous partial successes), and tool-specific code errors (e.g., malformed Boltz calls, RDKit conversion issues).
📜Paper: https://t.co/8WGtoutKjL
#LLM #DrugDiscovery #MedicinalChemistry #Cheminformatics #Benchmark #Agents #ADMET #StructureBasedDrugDesign #FragmentBasedDrugDiscovery #ComputationalBiology
A critical initialization for biological neural networks
Spontaneous brain activity is often treated as noise: the background hum of a nervous system waiting for a task. But large-scale recordings in mice have shown something more structured. Even in darkness, without explicit stimuli, thousands of neurons display coordinated activity patterns that extend across the brain and persist far longer than the fast biophysical timescales of individual neurons.
Marius Pachitariu and coauthors ask a simple question: could this macroscopic structure emerge from a simple kind of network initialization?
Their answer connects neuroscience, random matrix theory and machine learning. They model spontaneous neural activity as linear dynamics governed by a random connectivity matrix, stabilized by a global inhibitory-like normalization. When this matrix is symmetric and critically normalized, with its largest eigenvalue very close to one, the network naturally produces high-dimensional activity modes with a power-law covariance spectrum.
This is not just a mathematical curiosity. The same spectral structure appears in large-scale mouse recordings from cortex and brainwide Neuropixels data, with power-law exponents around 0.7–0.85. Hippocampal CA1 is the striking exception: its activity looks less correlated, closer to an efficient, high-capacity code for information storage.
The ML perspective is especially interesting. In artificial neural networks, initialization is often treated as a technical detail: Xavier, He, orthogonal schemes, and so on. But this paper reframes initialization as a computational substrate. A critically initialized recurrent system can generate slow, global, high-dimensional modes before task-specific learning. In simulations, these dynamics support time-dependent computations, including zero-shot working memory tasks.
The biological implication is powerful: spontaneous activity may not be random noise, but a preconfigured dynamical scaffold on which learning and computation can operate. The brain may start from an initialization already close to useful temporal memory, with learning then shaping readouts or task-specific pathways.
For R&D teams building ML systems in drug discovery, materials development, energy research or biotechnology, the lesson is broader than neuroscience. Initialization, architecture and dynamics define what kinds of scientific signals a model can preserve, combine and retrieve before training. In applied research pipelines where data are scarce, noisy and time-dependent, designing the right dynamical substrate may be as important as choosing the loss function.
Source: Pachitariu et al., Nature (2026) — CC BY 4.0 | https://t.co/oE37FfYmKc
What’s inside the PDBe MCP package?
⚡ API Server
Live structural biology data via REST-powered MCP tools
🔍 Search Server
Schema-aware Solr search across PDBe
🌐 Graph Server
Neo4j schema exploration + Cypher generation
One install.
Three servers.
Unlimited workflows
#MCP
We're pleased to share our first @Nature paper: Robin is the first multi-agent system for discovery in biology that integrates novel hypothesis generation with experimental data analysis in one continuous workflow.
In this study, our team, including ophthalmologist @agreeb66, applied Robin to dry age-related macular degeneration, a leading cause of irreversible sight loss with limited treatment options. The system proposed drug-repurposing hypotheses, which were then tested experimentally in the lab.
Robin developed the experimental strategy for therapeutic hypothesis generation, proposed follow-up experiments, and extracted actionable insights from the resulting data, including validation in primary human retinal pigment epithelium (RPE) stem cells.
Robin also proposed a mechanism of enhancing RPE phagocytosis by modulating the cells circadian rhythm using an experimental drug, KL001, that has never before been used in humans or proposed for AMD. To our knowledge, this mechanism had not previously been proposed.
This work points to the future of AI-enabled science: systems that connect insights across fields, surface new mechanisms, and turn existing knowledge into testable hypotheses.
It also represents the broader opportunity FutureHouse is building toward: AI that helps science cross disciplinary boundaries and move from literature to experiment to discovery.
https://t.co/4FhmTo8nRH
Lots of news today. Don't have energy to write a hit tweet, so here's list
1. Our work on doing lab-in-the-loop with agents was published in Nature
2. We announced our first research partnership with a pharmaceutical company
3. We made new persistent code-writing agent