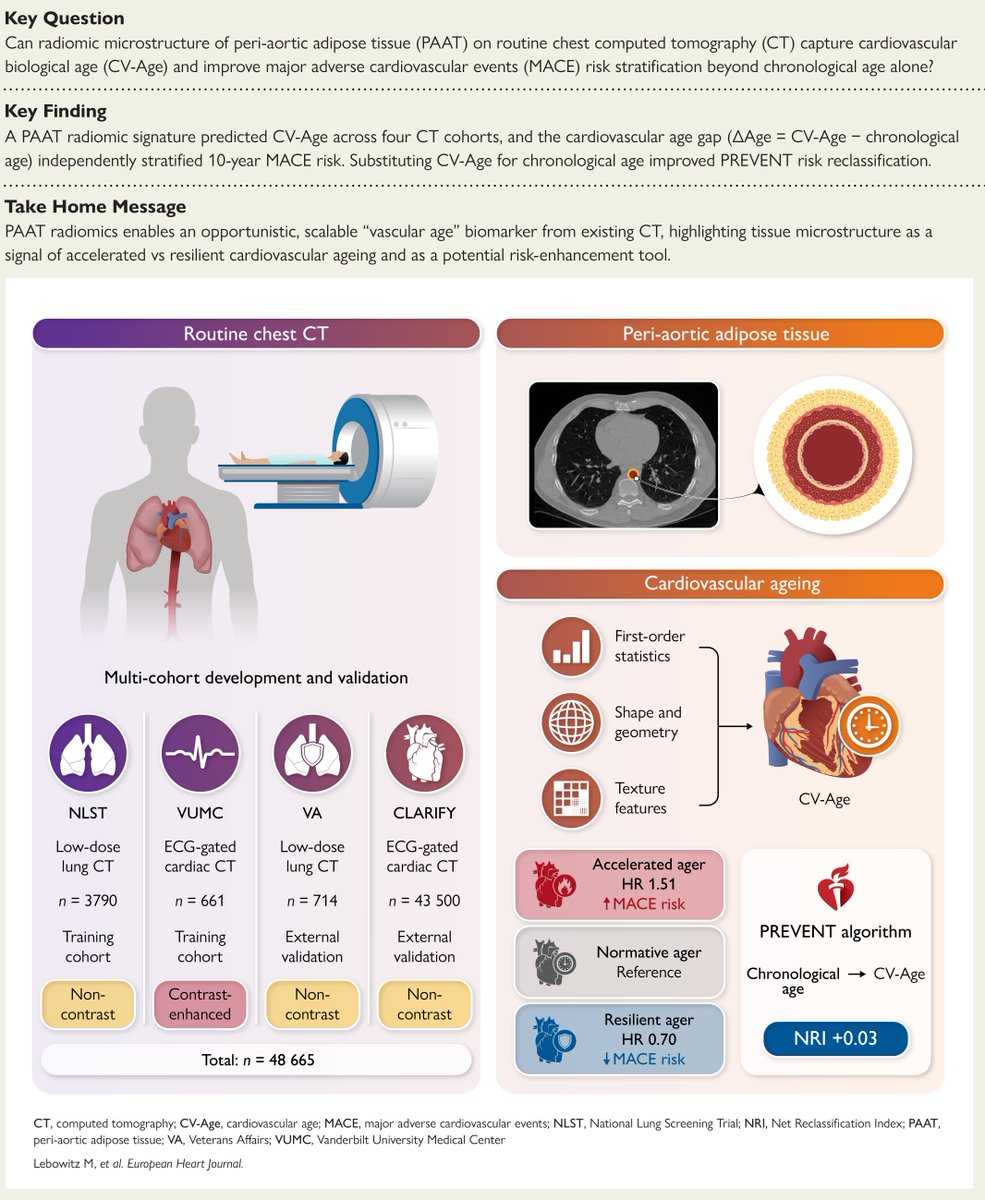
Your arteries have an age of their own and routine chest CT already knows it. An AI radiomic read of peri-aortic fat yields a cardiovascular age that beats the calendar. Raed more in #EHJ.
https://t.co/inUFhxp0qJ
#cardiotwitter@ESC_Journals@escardio
The science of human aging is flourishing, perhaps best exemplified by remarkable advances in organ and cellular clocks, tracked from proteins in the blood. These clocks tell us about the pace of aging within an individual and are linked to healthspan, longevity, and diseases.
@wysscoray and I reviewed the field of biological clocks, published today @NatureMedicine
free access
https://t.co/2PDZOgUmLu
Delayed gastric emptying appears to contribute only modestly to the appetite-suppressing effects of GLP-1 receptor agonists, with central mechanisms playing a more dominant role.
https://t.co/g4lmD6Yzl3
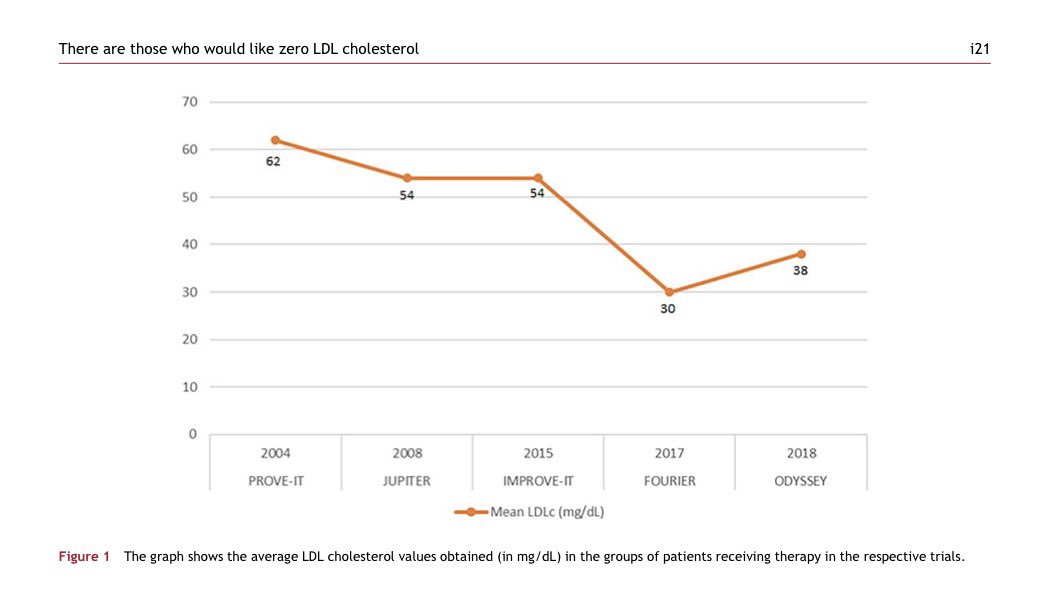
👉There are those who would like zero LDL cholesterol
🤔How low should LDL-C go?
For years we asked whether LDL-C could be too low.
Today, the evidence points in the opposite direction.
👉LDL-C is causal, not just associated with atherosclerosis.
👉Every additional reduction in LDL-C translates into additional cardiovascular risk reduction.
👉Clinical trials with statins, ezetimibe and PCSK9 inhibitors consistently show that lower is better.
👉Patients achieving LDL-C <20 mg/dL continue to derive cardiovascular benefit without convincing evidence of excess neurocognitive events, diabetes, or other major safety concerns.
👉Biology supports this concept: peripheral tissues require only minimal circulating LDL-C for physiological cholesterol delivery.
☝️The real question is no longer “Can LDL-C be too low?”
It is: “Why are so many high-risk patients still above target?”
👉The greatest danger is not an LDL-C of 20 mg/dL. It is therapeutic inertia.
@society_eas@nationallipid
🔓🔗 https://t.co/KaMaB9e1SA
The 2026 CKM Syndrome Guideline outlines the four stages of CKM syndrome, highlights major risk factors & provides comprehensive recommendations for screening, prevention & treatment. Check out the top 5️⃣ key takeaways 👇.
🔎 Learn more: https://t.co/151GI2RUYc #CardiologyMag
Most aging research assumes a gradual decline. A longitudinal Stanford study tracking 108 people aged 25 to 75 found the opposite—two distinct waves of abrupt biomolecular change that hit around the mid-40s and early 60s.
The traditional damage-accumulation model treats aging as oxidative stress, DNA mutations, and cellular dysfunction slowly building up over decades. The quasi-programmed framework argues that biological pathways optimized for growth and reproduction become dysregulated after reproductive years, driving hyperactive mTOR signaling and chronic inflammation.
Both frameworks miss something critical. Aging isn't linear.
Researchers used multi-omics technologies—transcriptomics measuring gene expression, proteomics tracking protein abundance and function, and metagenomics characterizing gut microbiome composition—to monitor blood and microbiome samples at regular intervals. The goal was to identify non-linear transitions in biomolecular profiles that signal pivotal moments in the aging process.
They found two distinct waves.
The first wave arrives around the mid-40s. Lipid metabolism shifts. ApoA1, the protein that packages HDL cholesterol and removes excess lipids from arterial walls, declines. ApoB, the protein that packages LDL particles carrying cholesterol into tissues, rises. This creates an environment that promotes cardiovascular disease.
At the same time, senescent cells accumulate. These are dysfunctional cells that have exited the cell cycle but persist in tissues. They adopt a senescence-associated secretory phenotype, releasing pro-inflammatory cytokines, chemokines, and proteases. While senescence prevents damaged cells from proliferating, the chronic presence of SASP creates a self-reinforcing cycle—inflammation triggers tissue damage, which produces more senescent cells, which amplify inflammation.
This state of chronic low-grade inflammation is called inflammaging, and it becomes a hallmark feature of the mid-40s wave. It doesn't just correlate with age-related disease. It drives cardiovascular disease, type 2 diabetes, and neurodegeneration by creating systemic dysfunction that compounds over time.
The gut microbiome also shifts during this period. Beneficial bacteria decline. Harmful species increase. This dysbiosis triggers systemic inflammation and insulin resistance, further exacerbating metabolic syndrome risk. The microbiome changes aren't just correlates of inflammation—they contribute to it by disrupting gut barrier integrity and allowing bacterial endotoxins to leak into circulation.
The second wave hits around the early 60s. Immune function declines through a process called immunosenescence, where the immune system loses its ability to mount effective responses to infections while simultaneously maintaining chronic inflammation. The result is a system that's both less responsive to pathogens and more inflammatory at baseline.
Carbohydrate metabolism deteriorates. Insulin resistance becomes more pronounced due to impaired glucose uptake in muscle and adipose tissue. Blood sugar rises. Metabolic syndrome risk escalates. Cardiovascular risk compounds.
Kidney function declines measurably. Glomerular filtration rate drops. Serum creatinine levels rise. The kidneys lose their ability to efficiently filter waste and maintain fluid balance, creating additional strain on cardiovascular and metabolic systems that are already compromised.
The gut microbiome undergoes further dysregulation during the second wave. Microbial diversity declines. Beneficial bacteria like Bacteroidetes and Firmicutes decrease. Harmful bacteria like Proteobacteria increase, creating a pro-inflammatory environment. Gut barrier dysfunction worsens, allowing lipopolysaccharides to leak into the bloodstream and trigger systemic inflammation that contributes to insulin resistance and neurodegeneration.
The wave-based model aligns with the quasi-programmed aging framework, which argues that aging results from the dysregulation of biological programs originally optimized for development and reproduction. This concept overlaps with antagonistic pleiotropy—the evolutionary theory that pathways conferring early-life benefits become detrimental later.
mTOR and the GH/IGF-1 axis are examples. These pathways drive growth and development during youth. They become maladaptive with age, promoting cellular dysfunction, inflammation, and tissue degeneration. Aging isn't random damage accumulation. It's the continuation of intrinsic biological programs operating in an environment they weren't optimized for.
This creates intervention opportunities. Rapamycin, an mTOR inhibitor, mitigates overactivation of growth pathways, promotes autophagy, and reduces inflammation. It's been shown to extend healthspan and lifespan across species by resetting dysregulated developmental programs.
Strategies that dampen the GH/IGF-1 axis—metformin, canagliflozin, caloric restriction—slow growth processes that accelerate aging and reduce risk of metabolic and degenerative diseases. These interventions target aging at its developmental roots, addressing the programs that drive dysfunction rather than just treating downstream damage.
The wave-based model highlights the need for precision health strategies tailored to the biological changes occurring at specific life stages. Biomarker-driven assessments can identify critical risk factors during the mid-40s and early 60s waves, allowing for targeted interventions—rapamycin protocols, metabolic optimization, microbiome restoration—before age-related diseases become clinically apparent.
The decisions made during the fourth and fifth decades about metabolic health, inflammation management, and microbiome composition may determine whether the transitions at 45 and 60 represent mild inflection points or catastrophic shifts that compound into chronic disease during the seventh and eighth decades.
Aging isn't a gradual decline. It's a series of abrupt, systemic transitions at predictable life stages. The mid-40s and early 60s represent critical windows where biological programs shift from maintaining homeostasis to driving dysfunction. The question is whether interventions targeting these transitions during the wave periods can prevent the compounding damage that defines late-life disease.
Llevamos años obsesionados con bajar los triglicéridos. Nueva revisión ➡️los TG no son directamente aterogénicos. Son el marcador de las lipoproteínas remanentes ricas en TG, que sí entran en la pared arterial. 🫀
Lo importante no es la cifra, es la carga de partículas apo B
@ESC_Journals
🔗 https://t.co/PfieuYrQSa
How many years until the guidelines consider CCTA as the preferred imaging tool to assess for the presence of disease, risk stratification and response to treatment?
#Perspective: CAC scanning remains the preferred imaging tool for cardiovascular risk stratification in most asymptomatic patients. CCTA should be reserved for select high-risk populations. https://t.co/DjPf3fr0d6
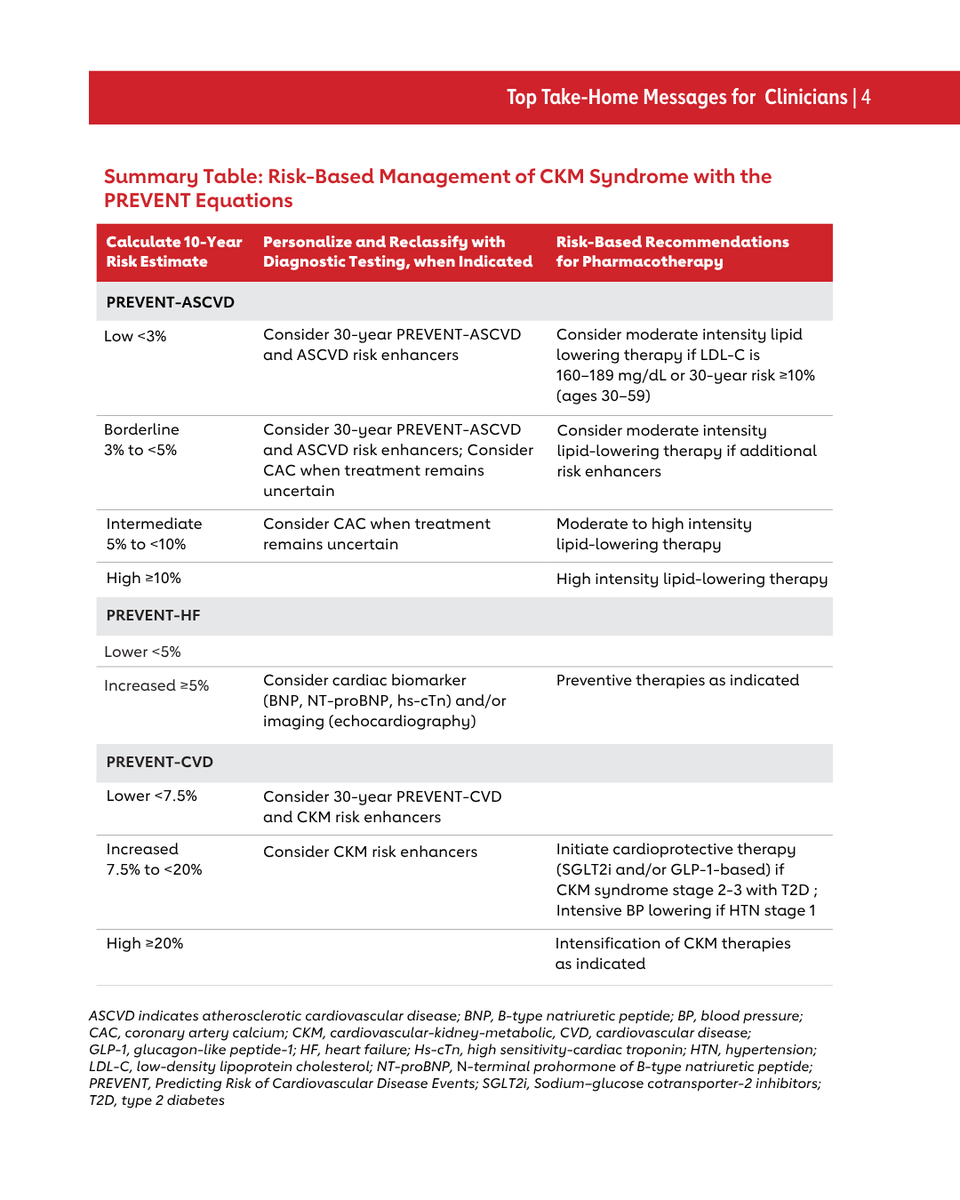
CKM Syndrome and PREVENT: Top Things to Know
The 2026 Guideline on the Management of CKM Syndrome was recently published.
Individuals at risk for cardiovascular disease (CKM syndrome stage 0-3) should have their risk quantified with the PREVENT (Predicting Risk of cardiovascular disease EVENTs) equations to estimate 10- and 30-year risk for atherosclerotic cardiovascular disease, heart failure, and total CVD.
PREVENT estimates inform CKM syndrome staging, with ≥20% predicted 10-year CVD risk serving as one criterion for CKM syndrome Stage 3. A ≥7.5% predicted 10-year CVD risk further informs the prioritization of pharmacotherapies
👉Lifetime LDL-C Exposure: The Forgotten Determinant of Atherosclerotic Risk
📍LDL-C is not just a number—it’s cumulative exposure over time.
📍Think beyond LDL-C levels. Think plaque-years.
📍The earlier you lower LDL-C, the greater the lifetime benefit.
📍Starting treatment at 45 is better than never—but starting at 30 changes the trajectory.
📍Every year with elevated LDL-C adds to the arterial burden. Those years cannot be erased.
📍Prevention is not about treating cholesterol. It’s about preventing decades of plaque accumulation.
📍The most powerful lipid-lowering therapy isn’t necessarily the strongest drug—it’s early intervention.
☝️Lower LDL-C. Earlier. Longer.
@society_eas@nationallipid
Sharing a slide from my recent talk on the future of obesity pharmacotherapy at the 34-lea Congres Național al Societății Române de Endocrinologie in București, România, illustrating the comparative effectiveness of current and emerging obesity medications.
Clopidogrel monotherapy was associated with lower risks of MACE and major bleeding compared with aspirin monotherapy among stable patients post #PCI, according to a nationwide cohort study published in #JACCAsia.
Learn more: https://t.co/QDar4mTyFP #CardioX@JACCJournals
In the latest meta-analysis, semaglutide is deemed the preferential GLP-1 RA in type 2 diabetes and chronic kidney disease.
Cardiorenal benefits appear consistent irrespective of background SGLT2 inhibitor use, consistent with guideline-recommended multi-pathway pharmacotherapy.
Thirteen RCTs, 97 428 participants
GLP-1 RAs ⬇️ MACE by 16%
⬇️ composite kidney endpoint by 21%
⬇️ Kidney failure risk by 28% (HR 0.72)
⬇️ UACR by 26%
Link: https://t.co/ee5cFf4JiC
In 773 pts from the PARADIGM registry, higher epicardial adipose tissue on #yesCCT was independently linked to coronary plaque progression and worse 10-yr #MACE-free survival, pointing to EAT as a potential marker for risk stratification. https://t.co/fVbEasYHU4 #JACCIMG
Apitegromab, a selective antimyostatin monoclonal antibody targeting muscle preservation, added to tirzepatide reduced muscle loss by ~55% over 24 weeks without affecting overall weight loss in the phase 2 EMBRAZE trial.
https://t.co/jDqTipQrZa
“Treat the plaque. Not the probability of finding one.*”
“If total plaque volume is already one of the strongest predictors of outcome, improving how we measure plaque—not just stenosis—may be where the next leap in preventive cardiology occurs.”
I believe that leap is already here for primary care and preventive medicine.
#CardiacCT #CCTA #Atherosclerosis #PreventiveCardiology #TreatThePlaque #CardiovascularImaging #PrecisionMedicine
@PHImaging
We Keep Predicting Risk. The Plaque Already Knows.
For decades, preventive cardiology has revolved around estimating the probability of obstructive CAD.
Age.
Sex.
Symptoms.
Risk factors.
This study asks a different question:
**What if the best predictor isn't the probability of disease... but the amount of disease already present?**
Using AI-based quantitative CCTA in more than **6,000 symptomatic patients**, the CONFIRM2 investigators found that **total plaque volume (TPV)** was the main driver of future cardiovascular events, regardless of whether patients had a very low, low, or moderate pre-test likelihood of obstructive CAD.
The numbers are striking.
At 4 years, MACE rates increased from:
- **1.4%** with no plaque
- **3.3%** with minimal plaque
- **12.6%** with TPV 250–750 mm³
- **18.6%** with TPV >750 mm³
And these event rates were remarkably consistent across all clinical likelihood categories.
That should make us pause.
For years we have refined clinical scores to estimate the likelihood of finding obstructive stenosis.
Meanwhile, the **actual burden of atherosclerosis** appears to carry far more prognostic information.
## The Bigger Message
This is another piece of evidence supporting a shift that has been emerging for years:
**Risk factors don't cause events.**
**Plaques do.**
Risk factors help us estimate who *might* have disease.
CCTA allows us to measure who *already has it*.
Those are fundamentally different questions.
## My Take
This paper is not really about AI.
AI simply makes comprehensive plaque quantification scalable.
The real story is that cardiovascular prevention is gradually moving away from a **risk-factor paradigm** toward a **disease-based paradigm**.
The artery has become the biomarker.
And once you can quantify the entire atherosclerotic burden, it becomes increasingly difficult to argue that stenosis should remain the center of the conversation.
## Where PCCT Comes In
This study used conventional CCTA.
Now imagine combining this biological approach with Photon Counting CT.
- Higher spatial resolution.
- Better characterization of non-calcified plaque.
- Improved detection of subtle high-risk features.
- More reproducible plaque quantification.
If total plaque volume is already one of the strongest predictors of outcome, improving how we measure plaque—not just stenosis—may be where the next leap in preventive cardiology occurs.
**Treat the plaque. Not the probability of finding one.**
#CardiacCT #CCTA #Atherosclerosis #PreventiveCardiology #TreatThePlaque #PhotonCountingCT #PCCT #ArtificialIntelligence #CardiovascularImaging #PrecisionMedicine
💉 Survodutide, a dual GLP-1/glucagon agonist, with Phase 3 results specifically in those with metabolic-associated steatotic liver disease (MASLD).
⬇️ 84% of survodutide group met primary endpoint of >30% liver fat content reduction by MRI, compared to 24% of placebo.
⬇️ Survodutide group had 12% weight reduction vs. 1% with placebo.
🔑 Glucagon agonism likely has direct liver benefits, independent of weight loss
📊 SYNCHRONIZE-MASLD published in Nature Medicine: https://t.co/ZOJ1HILgAH
The effect of current anti-amyloid drugs vs Alzheimer's disease is "trivial" for cognitive function or dementia severity, not clinically meaningful, from a systematic @cochranecollab review of 17 trials, >20,000 patients
https://t.co/9A6PeZOyAt