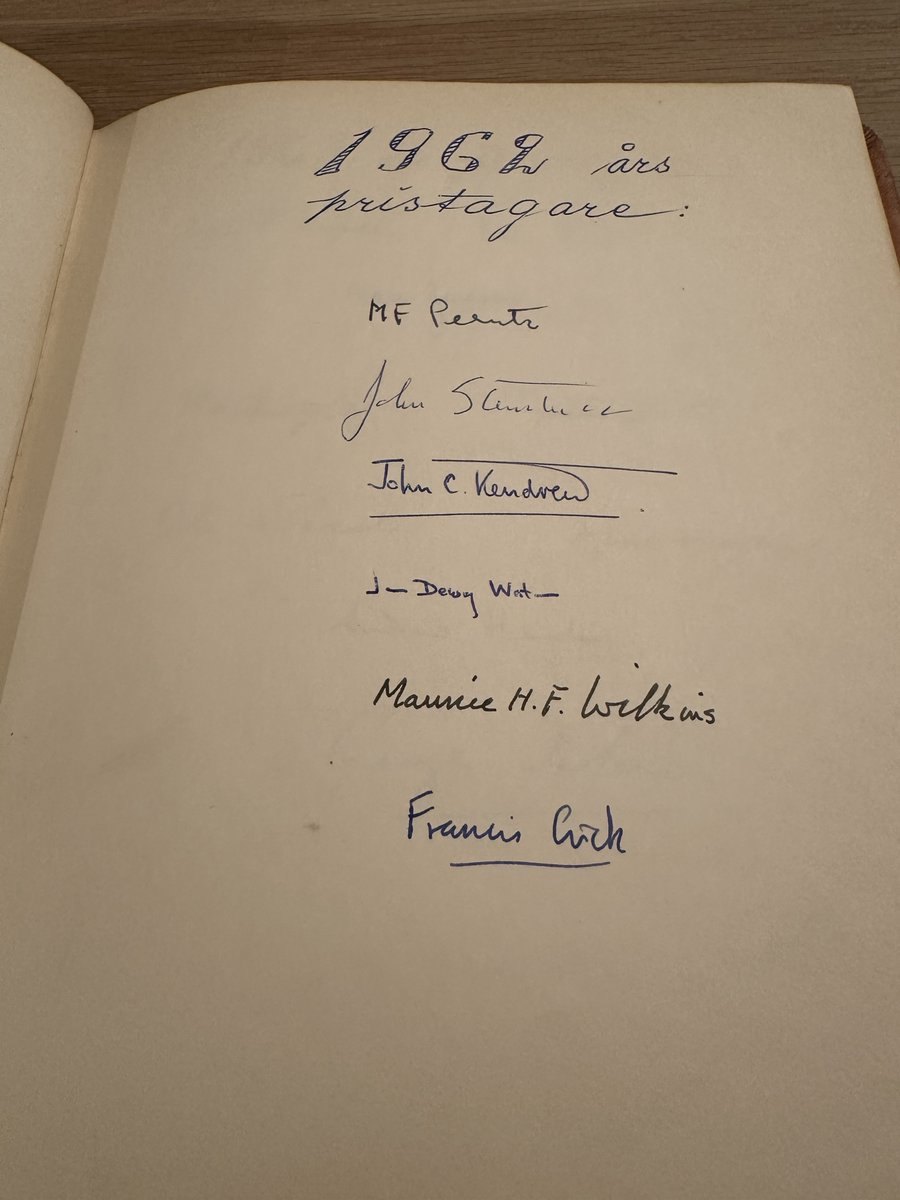Advances are being made to rationally design and develop cereblon-based molecular glue degraders - here's a perspective on their property-based optimization https://t.co/AzTZx18jcx
Glecirasib, a potent and selective covalent KRAS G12C inhibitor exhibiting synergism 2 with cetuximab or SHP2 inhibitor JAB-3312 https://t.co/h99wbdjfvN
Today we report in @NatureMedicine the development of a base editing strategy for prion disease, currently a fatal and rapidly progressing neurogenerative condition with no effective treatment. @PrionAlliance@DevermanLab (1/13)
https://t.co/jYVDj7JPBZ
CovCysPredictor: Predicting Selective Covalently Modifiable Cysteines Using Protein Structure and Interpretable Machine Learning https://t.co/pu2VUn8w9t
#compchem If your are interested by Karplus' scientific life, here is an interesting read: Spinach on the Ceiling: A Theoretical Chemist’s Return to Biology.
https://t.co/iR2qlGTBvL
It is very sad to learn that Martin Karplus passed away on Dec 28 at age 94. He was a scholar and pioneering chemical physicist with great contributions in many areas including molecular dynamics. We interacted many times starting from my graduate time at Harvard. His legacy is reinforced by the large number of coworkers who have also done much to advance computational science.
Chemoselective Stabilized Triphenylphosphonium Probes for Capturing Reactive Carbonyl Species and Regenerating Covalent Inhibitors with Acrylamide Warheads in Cellulo https://t.co/cFl6ArLecG
An incredible experience beyond imagining to get to sign the great Nobel book, alongside many of my all-time scientific heroes: Einstein, Curie, Bohr, Planck, Schrodinger, Crick, Feynman…
This summary of major clinical updates from November covers FDA approvals, the initiation of new clinical trials, notable trial outcomes for key drug candidates, and news on paused and discontinued trials and programs.
Read more here: https://t.co/mkKb6CIxkh
You may have seen a recent pre-print [1] from Jain et al. with strongly worded claims against the experimental results in our DiffDock paper [2]. We initially declined to respond as we saw that this preprint contained falsehoods, misleading comparisons, seemingly deliberate omissions, and was written in a tone not intended as a serious research paper. Unfortunately, the preprint has now received undue attention on social media and beyond. After receiving inquiries for us to respond, we decided to do so.
First, we wish to emphasize that, similar to all of our papers, we published all the code and data. Doing so ensures that the work is always reproducible and any aspect of it can be openly assessed and critiqued by anyone. Further, we have openly acknowledged limitations in the original manuscript, and have addressed these limitations in follow-up papers [3,4,30]. We cherish all the constructive feedback we have received on our work and the collaborations that arose to improve the method.
The main false claim in Jain et al. is that the DiffDock paper poorly selected train/test splits and incorrectly ran baselines. As Jain et al. well know, neither the dataset split nor the contended baselines were introduced in the DiffDock paper: we used baselines and benchmarks established by prior peer-reviewed work [6,7], and these have been used in many other concurrent [8,9] and subsequent works [10,11,12, …] as well. We certainly agree that the specific benchmark did not sufficiently evaluate docking generalization, which is why we also reported performance on unseen receptors in the original paper [2, Table 7 and Figure 7] and we proposed better evaluations in later papers [3,4]. This conclusion was shared by many other groups in the field and has helped build better alternatives [13,4], which in turn has led to significant progress in the field over the past three years.
Below we further highlight some specific incorrect statements, misleading comparisons, and deliberate omissions in Jain et al.
1. Jain et al. use incorrect statements to bolster their points. For example, they claim "DiffDock made use of roughly 17,000 co-crystal structures for learning (98% of PDBBind version 2020” [1, abstract] while the total protein-small molecule structures in PDBBind v2020 is 19443 and the size of the training set was 16379, and thus 16379 out of 19443 used for training is 84.2%, not 98% that they also repeat in social media. The established test set was curated from the remaining complexes (beyond 84.2%) to avoid any ligand overlap with the training set.
2. Improving the benchmarks is a topic that we (along with the rest of the field) have spent a lot of effort on over the past three years [3, 4], building harder and larger test sets. Jain et al. were well aware of these efforts but decided to ignore them in their paper.
3. Many of the empirical results presented by Jain et al. are apples-to-oranges comparisons. In the DiffDock paper, we were very clear that the goal was blind docking where pockets are not given as input to the methods. We also clearly stated that “search-based methods work best when given a starting binding pocket to restrict the search space” [2, page 8]. Regardless, four out of the five sets of benchmarking results in Jain et al. are misleadingly comparing a method that takes as input the pocket to DiffDock which does not.
4. In the only evaluation where all methods receive the same input information, Jain et al. exclusively use commercial non-academically accessible software. Jain et al. ran the Surflex-Dock docking pipeline in a blind setting and showed comparable performance. While we always welcome new methods and baselines, we feel that baselines in scientific literature should be accessible to researchers (not hidden behind a paywall) and open to scrutiny.
5. Jain et al. omit to mention that we introduced docking pipelines similar to what they advocate for already in the DiffDock paper [page 8 and appendix D.4]. We integrated open-source pocket-finding methods such as P2Rank [14] with open-source docking methods like GNINA [15] and showed that this indeed significantly improved classical docking methods for blind docking.
6. Jain et al. present, as novel, results that have been already highlighted by multiple works. In particular, they highlight the challenge of DiffDock to generalize well to unseen proteins. We reported performance on unseen receptors already in the original paper [2, Table 7 and Figure 7] and this was the reason for our follow-up paper [3] (as well as many others in the field) more than a year ago. Generalization to unseen proteins is a crucial challenge that the field has been collectively tackling with rapid progress annually. It is notable that Jain et al. did not compare to the state of the art as is customary in scientific practice, rather selecting a method from more than two years ago. For example, our works [3,30], all available openly, have already addressed the exact points they focus on.
7. Jain et al. appear to attack our paper as misleading practitioners. This seems like a strange claim against work that is all open, readily accessible, testable, and reproducible. We are aware of several companies having seen benefits by integrating DiffDock into their pipelines (many, for example, through the NVIDIA BioNeMo platform) and there are several papers highlighting biological discoveries where DiffDock played a part [for example 16,17,18,19,20,21,22,23,24,25,26,27,28,29].
8. Jain et al. do not acknowledge their apparent financial conflict of interest with the advocacy of their claims and results.
I would add that it is sad to see respected scientists like Derek Lowe propagating such false and misleading claims [5], without minimal due diligence such as reaching out to us for comment or independently verifying statements for a fair and accurate coverage.
[1] Jain, Ajay N. et al. “Deep-Learning Based Docking Methods: Fair Comparisons to Conventional Docking Workflows”. arXiv preprint (2024).
[2] Corso, Gabriele, et al. "Diffdock: Diffusion steps, twists, and turns for molecular docking." International Conference on Learning Representations (2023).
[3] Corso, Gabriele, et al. "Deep confident steps to new pockets: Strategies for docking generalization." International Conference on Learning Representations (2024).
[4] Durairaj, Janani, et al. "PLINDER: The protein-ligand interactions dataset and evaluation resource." bioRxiv preprint (2024).
[5] Lowe, Derek. “Computational Care”. In the pipeline, Science Commentary Blog (2024).
[6] Stärk, Hannes, et al. "Equibind: Geometric deep learning for drug binding structure prediction." International Conference on Machine Learning (2022).
[7] Lu, Wei, et al. "Tankbind: Trigonometry-aware neural networks for drug-protein binding structure prediction." Advances in neural information processing systems 35 (2022).
[8] Zhang, Yangtian, et al. "E3bind: An end-to-end equivariant network for protein-ligand docking." International Conference on Learning Representations (2023).
[9] Qiao, Zhuoran, et al. "State-specific protein–ligand complex structure prediction with a multiscale deep generative model." Nature Machine Intelligence 6.2 (2024).
[10] Lu, Wei, et al. "DynamicBind: Predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model." Nature Communications 15.1 (2024).
[11] Pei, Qizhi, et al. "Fabind: Fast and accurate protein-ligand binding." Advances in Neural Information Processing Systems 36 (2024).
[12] Huang, Yufei, et al. "Re-Dock: Towards Flexible and Realistic Molecular Docking with Diffusion Bridge." Advances in Neural Information Processing Systems 36 (2024).
[13] Buttenschoen, Martin et al. "PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences." Chemical Science 15.9 (2024).
[14] Krivák, Radoslav, and David Hoksza. "P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure." Journal of Cheminformatics 10 (2018).
[15] McNutt, Andrew T., et al. "GNINA 1.0: molecular docking with deep learning." Journal of Cheminformatics 13.1 (2021).
[16] Peh, GuangRong, et al. "Further Characterization of Fungal Halogenase RadH and Its Homologs." Biomolecules 13.7 (2023).
[17] Kim, Soo-Kyoung, et al. "Diribonuclease activity eliminates toxic diribonucleotide accumulation." Cell reports 43.9 (2024).
[18] Ross, Patricia A., et al. "Framework for exploring the sensory repertoire of the human gut microbiota." mBio 15.6 (2024).
[19] Zhovmer, Alexander S., et al. "Septins provide microenvironment sensing and cortical actomyosin partitioning in motile amoeboid T lymphocytes." Science Advances 10.1 (2024).
[20] Li, Jinmei, et al. "A dual MTOR/NAD+ acting gerotherapy." bioRxiv (2023).
[21] Koplūnaitė, Martyna, et al. "Exploring the Mutated Kinases for Chemoenzymatic Synthesis of N 4-Modified Cytidine Monophosphates." Molecules 29.16 (2024).
[22] Bu, Yingzi, et al. "A gastrointestinal locally activating Janus kinase inhibitor to treat ulcerative colitis." Journal of Biological Chemistry 299.12 (2023).
[23] Reilly, Charles B., et al. "Broad-Spectrum Coronavirus Inhibitors Discovered by Modeling Viral Fusion Dynamics." bioRxiv (2024).
[24] Esquea, Emily, et al. "Discovery of novel brain permeable human ACSS2 inhibitors for blocking breast cancer brain metastatic growth." bioRxiv (2023).
[25] Suryawanshi, Vikramsinh Sardarsinh, et al. "Machine learning-integrated and fingerprint-based similarity search against immuno oncology library for identification of novel ERK2 inhibitors." Structural Chemistry (2024).
[26] Xu, Jiangcheng, et al. "Developing Lead Compounds of eEF2K Inhibitors Using Ligand–Receptor Complex Structures." Processes 12.7 (2024).
[27] Baroni, Sara, et al. "The antipsychotic drug lurasidone inhibits coronaviruses by affecting multiple targets." Frontiers in Cellular and Infection Microbiology 14 (2024).
[28] B Fortela, Dhan Lord, et al. "Predicting molecular docking of per-and polyfluoroalkyl substances to blood protein using generative artificial intelligence algorithm DiffDock." Biotechniques 76.1 (2024).
[29] Miyairi, Yuichi, et al. "A class of chemical compounds enhances clustering of muscle nicotinic acetylcholine receptor in cultured myogenic cells." Biochemical and Biophysical Research Communications 731 (2024).
[30] Wohlwend, Jeremy, et al. "Boltz-1: Democratizing Biomolecular Interaction Modeling." bioRxiv (2024).
Super excited to preprint our work on developing a Biomolecular Emulator (BioEmu): Scalable emulation of protein equilibrium ensembles with generative deep learning from @MSFTResearch AI for Science.
#ML#AI#NeuralNetworks#Biology#AI4Science
https://t.co/yzOy6tAoPv