(1/2) scTranslator: Training on large-scale RNA data to impute protein abundance.
- pretrain on paired RNA+ protein in sc and bulk and predict on single-cell RNA-only data
- Genes are encoded by raw expression embedding combined with gene embedding
Happy to share that our study of co-differentiation of cardiomyocytes and endothelial cells from human iPSCs was published in Stem Cells https://t.co/RA7aTT8roA Excellent work by @Xu_Cao_PhD and @Siliegia and a great collaboration with the Orlova lab.
Thrilled to announce that our paper on XEN-enhanced gastruloids is now online https://t.co/BzVpcsduM3 . We hope you'll find it interesting. Thanks to everybody involved for all of the hard work!
How does a cell decide its own fate? Read more into this fascinating review by @SemrauLab and @Siliegia where they delve into a single-cell view of cell-type specification #BiochemSocTrans#openaccess https://t.co/PG8b5OdtaI
Our new paper from @SemrauLab on clusterability of scRNA-seq data is now published in Genome Biology! The new name is phiclust and the code is available at Github https://t.co/AqtH0CGshb.
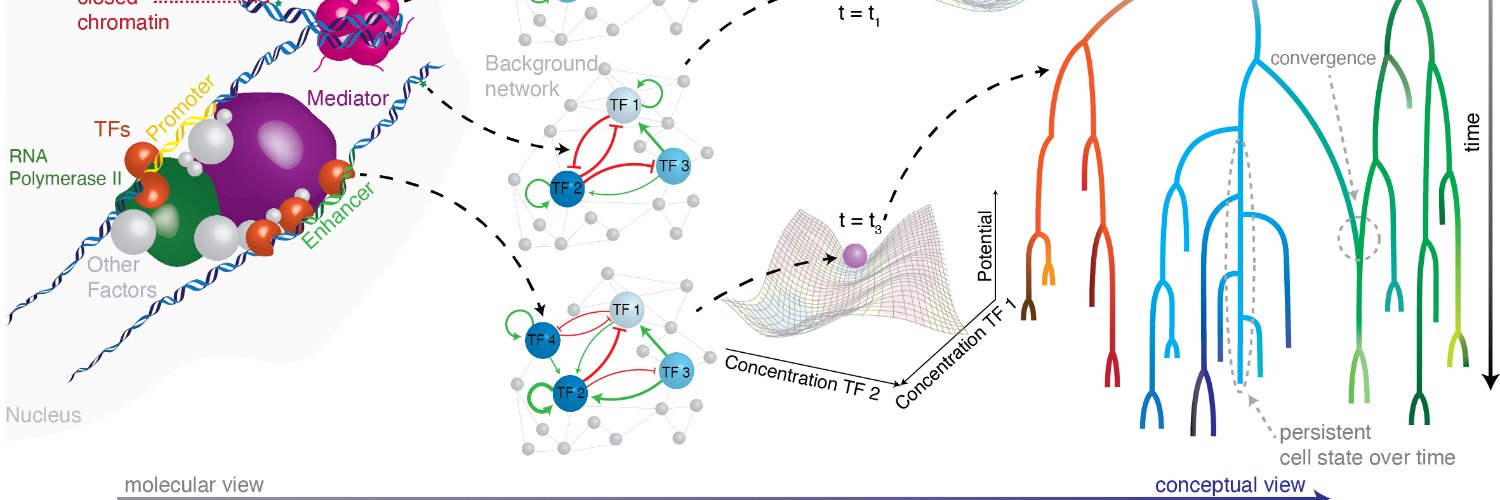
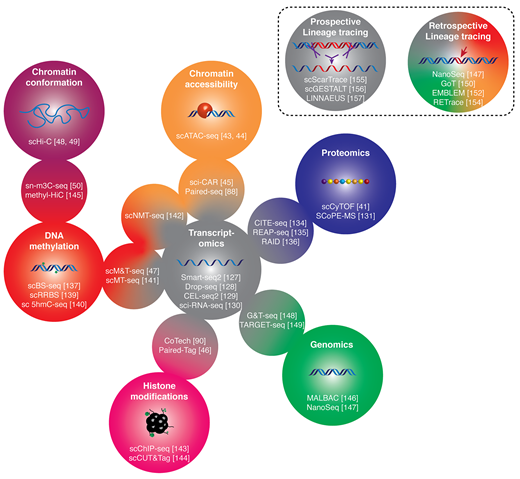
Are you interested in the decision-making of single cells? Check out our new review paper from @SemrauLab. We review the important molecular players and single-cell methods to measure them!
https://t.co/67B2zO1hWg
@CasualVariant@razoralign (5) It is not possible to recover the full signal matrix (without random noise) which is why we only calculate the distance from the measured expression matrix to the real (unobservable) signal.
@CasualVariant@razoralign (4) On the other hand, the low-rank approximation is not necessarily noise-free by the conventional definition of noise (random effects) but merely non-sparse.
@CasualVariant@razoralign (2) From that point of view the low-rank signal is also permitted to be sparse. We rather define a pure cell population by having only fluctuations in gene expression space that can be explained by randomness.
@CasualVariant@razoralign (1) Great question! There are two main differences:
We do not assume the noise matrix to be sparse, but only to be random. So, we disentangle random variations from deterministic ones, rather than sparse signals from low-rank signals.
Is a scRNA-seq cluster worth sub-clustering or are the fluctuations consistent with random noise? Check out SIGMA, our new clusterability measure for scRNA-seq data, purely based on first principles! By @SemrauLab https://t.co/ok08vbqtZT
Tomorrow, April 20 from 15.00h to 17.00h, scNL will be hosting our first meeting online! Join us to brainstorm about what scNL should become here: https://t.co/5gbaNSybbV