📣 new preprint multimodal atlas. Imaging + scRNA, 57M cells. 🧬🔬
Cells are complex dynamical systems — but most ways we measure them destroy them. We asked: how does live imaging compare to scRNA-seq, the field’s gold std?
The answer surprised us 🧵
https://t.co/RG0PZ1KTHW
We’re excited to share our latest publication in @NatComputSci “MetaSTAARlite: an all-in-one tool for biobank-scale whole-genome sequencing meta-analysis” 🧬
Sincere thanks to Yohhan Kumarasinghe (UNC, co-lead), Jacob Williams (NCI, co-lead), Yuxin Yuan, Wenbo Wang, @AlexiaDiasF, @AndrewHaoyu, @muzizimumu1, and the study participants from @uk_biobank and @AllofUsResearch.
Biobank-scale WGS/WES studies are transforming rare variant discovery, but pooled individual-level analyses across biobanks are often limited by data-sharing restrictions. MetaSTAARlite is designed to overcome this challenge by providing a scalable, resource-efficient, summary statistics–based pipeline for functionally informed rare variant meta-analysis across the coding and noncoding genome.
MetaSTAARlite provides an all-in-one workflow to:
• Generate resource-efficient study-specific summary statistics, including variant-level summary statistics + sparse LD matrices
• Perform functionally informed coding, noncoding, ncRNA, and custom-mask rare variant meta-analysis
• Dynamically incorporate multiple variant functional annotations to improve power and interpretation
• Exactly reconstruct the variance-covariance matrix of score statistics (referred to as the LD matrix), so meta-analysis results closely mirror pooled individual-level analysis
• Account for population structure and relatedness using sparse GRM / mixed-model framework
• Support conditional analysis, Manhattan/QQ plots, and analytical follow-up to identify annotations and variants driving associations
A key advantage of MetaSTAARlite is scalability. By leveraging sparse GRM and directly operating on sparse genotype dosage matrices, MetaSTAARlite greatly reduces runtime, memory, and storage. Benchmarking on UK Biobank WES data for TTN missense variants (the largest gene in the human genome) and total cholesterol phenotype:
• At n = 300K, MetaSTAARlite achieved 332× and 1,386× lower peak memory than MetaSTAAR and Raremetal2, respectively
• It also achieved 24× and 2,206× lower computation time than MetaSTAAR and Raremetal2
• At n = 446K with 22,994 variants, summary statistics generation finished in 48.82 seconds with <1 GB peak memory
Another important challenge in rare variant meta-analysis is the storage of LD matrices. MetaSTAARlite substantially reduces this burden. In a UK Biobank WGS total cholesterol benchmark, we randomly partitioned 190,110 participants into three studies and generated genome-wide summary statistics with 12 functional annotations. MetaSTAARlite required only:
• 0.48 GB total storage per mask for genome-wide coding meta-analysis summary statistics
• 1.67 GB total storage per mask for genome-wide noncoding meta-analysis summary statistics
Notably, the sparse LD matrices accounted for only 7.7% and 17.8% of the total storage for coding and noncoding analyses, respectively. This means that, in MetaSTAARlite, LD matrix storage is no longer a bottleneck for rare variant meta-analysis.
In UK Biobank WGS total cholesterol analyses, we compared MetaSTAARlite meta-analysis with pooled STAARpipeline analysis using the same individual-level data. The results were nearly perfectly concordant:
• Pearson r² > 0.999 for log10-transformed P values across genome-wide significant and suggestive masks
• 58 genome-wide significant coding associations
• 88 genome-wide significant noncoding associations
• Signals included known lipid biology: PCSK9, APOB, APOA5, LDLR and APOE clusters
We further applied MetaSTAARlite to cross-biobank meta-analysis of UK Biobank (in the Research Analysis Platform) and All of Us (in the Research Workbench) data for five traits: total cholesterol, height, eGFR, calcium and elevated LDL-C (a binary trait). These analyses included up to 692,445 diverse participants. Across these traits, MetaSTAARlite identified 165, 536, 117, 38 and 94 genome-wide significant coding associations, respectively, while keeping average peak memory below 1 GB.
For these cloud-based analyses, in the UK Biobank RAP, for example, the genome-wide summary-statistics generation per trait had theoretical costs of ~£3.60–£3.90 and actual costs typically below £7, never exceeding ~£8.20, for a total of 5 masks across the genome.
We hope MetaSTAARlite will make cross-biobank rare variant discovery more accessible, scalable and privacy-preserving for large WGS/WES consortia.
Software and tutorial are open source:
Paper: https://t.co/igDbXpx513
MetaSTAARlite: https://t.co/4QQ2ke3sCR
Tutorial: https://t.co/fGntgdVR4V
Manuscript code: https://t.co/s4wioFwH3W
🧬To better understand genome wide association studies we need to look beyond tissues and zoom in on individual cell types.
A remarkable @Nature study shows that eQTLs detected in specific cell types explain IBD GWAS signals better than eQTLs detected in pooled cells or broader cell populations.
The authors built IBDverse — large single-cell atlas based on scRNA-seq from blood, rectum and terminal ileum. The cohort included 421 people, including 125 patients with Crohn’s disease. After quality control, the analysis covered nearly 2.2 million single cells with matched genotypes from 396 people.
Cell type eQTLs were more often located far from transcription start sites, enriched in enhancers rather than promoters, less likely to regulate the nearest gene and more than 3.5 times more likely to colocalize with IBD GWAS loci than lower resolution eQTLs.
This matters because GWAS signals often sit in enhancers. Single cell data can therefore get closer to disease relevant biology.
The authors nominated likely effector genes for 180 of 321 known IBD loci (56%). For 74 loci this was the first effector gene nomination compared with previous eQTL annotations in Open Targets Genetics.
The study also reframes IBD as more than an immune disease. If the epithelial barrier renews or repairs poorly, the gut becomes more vulnerable. Inflammation may then be not only a cause of tissue damage but also a consequence of weak tissue repair.
#IBD #GWAS #scRNA #eQTL #colocalization
https://t.co/tIkMnDYlCT
Exciting breakthrough technology from the lab, now live in @CellCellPress ! Instead of cutting the genome where proteins bind (e.g., Cut&Tag), D&D-seq scars the DNA with a deaminase, allowing single cell genome mapping of TFs and chromatin remodellers!
🧬 A beautiful @NatureGenet paper on how to handle polygenic risk more carefully in family-based data.
The core idea is to compare a child not with random people, but with the genetic set they could have inherited from their own parents. This better protects against population stratification, geography, social structure, and assortative mating.
The authors introduce PGS-TRI, a method that estimates not only the direct effect of the child’s PGS, but also gene–environment interactions and asymmetric indirect effects of maternal and paternal genetics.
For the educational-attainment simulations, the authors used UK Biobank to create a realistic confounded setting with geography, BMI, and assortative mating. Standard regression became biased, while PGS-TRI stayed well calibrated.
In real autism trios, the method found a direct polygenic effect close to previous case–control estimates. It also showed that the score’s effect declines smoothly with genetic distance from the European training population.
https://t.co/PV9vmyUdVZ
#Trio #PopulationStratification #Autism #PRS #PGS
Drop it like its 🔥 ... Hot of the press.. #spatialomics review of all things #computation
For pros or new adopters to #spatialtranscriptomic & #spatialproteomic analyses.
Our comprehensive guide to software tools currently used in the field. https://t.co/G4iZce4R0r
1/5
Cell identity is written in the proteome, not in the DNA, and not always in the RNA. Out on bioRxiv today: The first cell type-resolved, MS-based proteomic atlas of the human body.
https://t.co/5RJ0nVoQ81
Excited to share Decima, out now in @naturemethods! 🎉
Existing seq-to-function models predict bulk expression. Decima goes further: it predicts gene expression in specific cell types and disease states from DNA sequence alone — trained on 22M+ single cells.
Applications: cis-regulatory mechanisms, cell-type-resolved variant effect prediction, and designing context-specific regulatory DNA
This preprint and accompanying browser provide another fantastic resource for exploring causal biology, with genotype–phenotype associations for both common and rare variants across 3,602 traits in the All of Us cohort (N=392,030)!
🧬Another ~1% step toward closing the missing heritability gap.
When we talk about genetic variants, we usually mean single nucleotide substitutions, or SNPs. But every human genome also carries ~25,000 structural variants, including insertions, deletions, inversions and other rearrangements of DNA segments ≥50 bp. These variants are one plausible contributor to missing heritability: the gap between heritability estimated from family and twin studies and what we can explain from measured genetic variants.
In this @NatureGenet paper, the authors built a catalogue of 171,233 high-quality structural variants from long-read, haplotype-resolved assemblies of 241 genomes. They then created ImputeSV, a pipeline to infer SVs from SNP data, and applied it to UK Biobank. The result: 54,578 common SVs imputed in 456,643 participants of European ancestry.
For some biomarkers, the contribution was substantial. In a joint model with small variants, SVs explained ~14% of variance in total bilirubin and ~12% in lipoprotein(a).
A particularly elegant part is variable number tandem repeats, or VNTRs. These behave like a genetic volume knob. For example, a VNTR in GGT1 showed a length-dependent association with γ-glutamyltransferase, a liver enzyme used as a marker of hepatobiliary function.
Overall, the authors found 17,335 SV–trait associations, including 958 loci unlikely to be explained by nearby SNPs alone.
Importantly, the authors released an imputation resource. This means existing SNP-based cohorts can now be re-analysed for SVs and VNTRs without long-read sequencing every participant.
https://t.co/pX1uT3qBLP
#GWAS #StucturalVariants #Imputation #VNTR
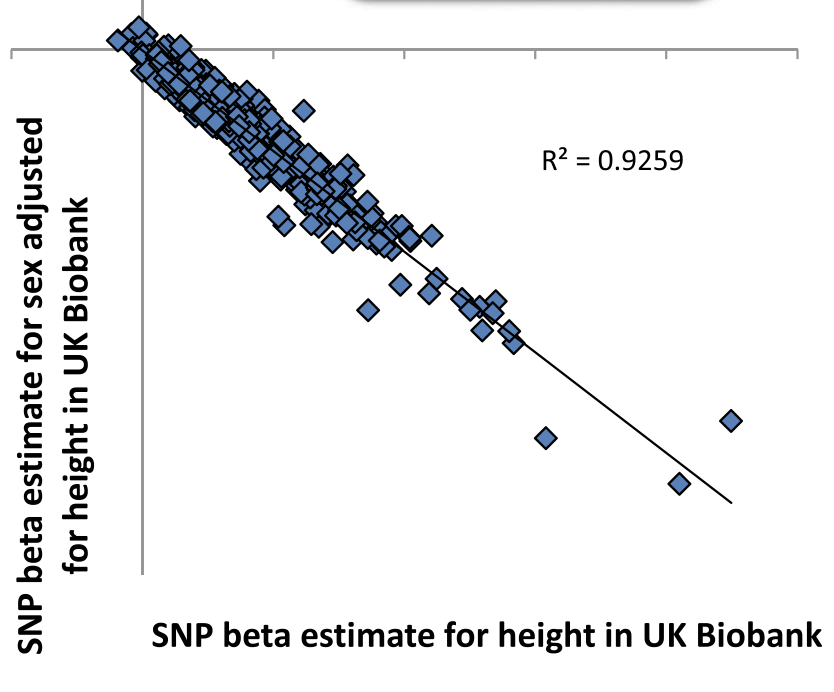
Collider bias can also influence genetic associations.
In a nice illustration, if we take height-raising SNPs and test their effects on sex (which should be null), then adjust for height, we obtain spurious associations with female sex.
Conditioning on a collider (height), associated with both the tested SNPs and sex, induces an association between the two.
A new amazing resource for drug target explorations with genomic data was released today @Nature:
A massive meta-analysis GWAS for 249 NMR-quantified metabolites in UK Biobank and Estonian Biobank across 619,372 individuals👇
SV-GWAS is highly accessible now! Our new paper in @NatureGenet shows how HiFi long-read assemblies let us repurpose SNP-based GWAS data to impute structural variants (SVs) to interrogate their role in human complex traits and diseases. @WeiyangBai https://t.co/zSB90E3at0
Very excited about this preprint that we just posted! It introduces the Genomic-Relatedness Matched Association (GRMA) study. It’s an extension to family-based GWAS that uses extended relatives beyond only siblings in diverse-ancestry data with very little bias.