𝐁𝐫𝐞𝐚𝐤𝐢𝐧𝐠 𝐭𝐡𝐞 𝐒𝐩𝐞𝐞𝐝 𝐋𝐢𝐦𝐢𝐭 𝐢𝐧 𝐍𝐞𝐮𝐫𝐚𝐥 𝐍𝐞𝐭𝐰𝐨𝐫𝐤 𝐌𝐨𝐥𝐞𝐜𝐮𝐥𝐚𝐫 𝐃𝐲𝐧��𝐦𝐢𝐜𝐬!
I am thrilled to share our latest paper published in the Journal of Chemical Theory and Computation (JCTC) @JCIM_JCTC : "𝐅𝐚𝐬𝐭𝐞𝐫 𝐌𝐨𝐥𝐞𝐜𝐮𝐥𝐚𝐫-𝐃𝐲𝐧𝐚𝐦𝐢𝐜𝐬 𝐰𝐢𝐭𝐡 𝐍𝐞𝐮𝐫𝐚𝐥 𝐍𝐞𝐭𝐰𝐨𝐫𝐤 𝐏𝐨𝐭𝐞𝐧𝐭𝐢𝐚𝐥𝐬 𝐯𝐢𝐚 𝐃𝐢𝐬𝐭𝐢𝐥𝐥𝐞𝐝 𝐌𝐮𝐥𝐭𝐢𝐩𝐥𝐞 𝐓𝐢𝐦𝐞-𝐬𝐭𝐞𝐩𝐩𝐢𝐧𝐠 𝐚𝐧𝐝 𝐍𝐨𝐧𝐜𝐨𝐧𝐬𝐞𝐫𝐯𝐚𝐭𝐢𝐯𝐞 𝐅𝐨𝐫𝐜𝐞𝐬".
Link to the paper: https://t.co/0R8VT0iC2L
Link to the updated preprint (#openaccess): https://t.co/SIbycvnEkz
In this work, we introduce DMTS-NC, a model-agnostic acceleration strategy that bridges the performance gap between high-accuracy Neural Network Potentials (NNPs) and traditional classical force fields. By distilling large foundation models (like FeNNix-Bio1 and MACE-OFF23) into a lightweight architecture that directly predicts nonconservative forces, we bypass the computationally expensive backpropagation step entirely. To ensure exceptional simulation stability without requiring tedious, system-specific fine-tuning, the architecture strictly enforces key physical priors like rotational equivariance and net-zero atomic force cancellation. The results are a game-changer for high-throughput simulations: DMTS-NC delivers a 15% 𝐭𝐨 30% 𝐬𝐩𝐞𝐞𝐝𝐮𝐩 𝐨𝐯𝐞𝐫 𝐩𝐫𝐞𝐯𝐢𝐨𝐮𝐬 𝐜𝐨𝐧𝐬𝐞𝐫𝐯𝐚𝐭𝐢𝐯𝐞 𝐌𝐓𝐒 𝐟𝐫𝐚𝐦𝐞𝐰𝐨𝐫𝐤𝐬 𝐚𝐧𝐝 𝐮𝐩 𝐭𝐨 𝐚 5.6𝐱 𝐚𝐜𝐜𝐞𝐥𝐞𝐫𝐚𝐭𝐢𝐨𝐧 𝐨𝐯𝐞𝐫 𝐬𝐢𝐧𝐠𝐥𝐞-𝐭𝐢𝐦𝐞-𝐬𝐭𝐞𝐩 𝐦𝐞𝐭𝐡𝐨𝐝𝐬. Furthermore, by pairing this approach with Hydrogen Mass Repartitioning (HMR) and High Hydrogen Friction (HHF), we successfully extended outer time steps up to 10 fs while maintaining flawless thermodynamic and free-energy accuracy.
Implemented in Tinker-HP @TINKERtoolsMD !
Huge congratulations to the team at @qubit_pharma and Sorbonne Université/CNRS: @NicolaiGouraud, @comecattin, Olivier ADJOUA, Louis Lagardère, and @Thomas__Ple !
Funding from @ERC_Research(project EMC2) and supercomputing time @Genci_fr .
#MolecularDynamics #MachineLearning #compchem #compbio #AI4Science #foundationmodels #FeNNixBio1
#quantumcomputing New group preprint: "Experimental Realization of the Markov Chain Monte Carlo Algorithm on a Quantum Computer".
https://t.co/NSGNw3gTin
In this work, we experimentally use encodings of Markov chains to prepare quantum states and run a quantum Markov Chain Monte Carlo algorithm (qMCMC) on Quantinuum's H2 and Helios quantum computers. We demonstrate that it is possible to obtain accurate results on current Noisy Intermediate Scale Quantum (NISQ) hardware, operating directly on the physical qubits.
Great work by B. Claudon @qubit_pharma and nice collaboration with S. Ramos-Calderer @quantumlah.
#compchem Our paper in J. Phys. Chem. Lett. (@JPhysChem): "Accelerating Molecular Dynamics Simulations with Foundation Neural Network Models using Multiple Time-Step and Distillation" made it to one of the covers! https://t.co/umkOcE0vvT
💫 As promised, we just released on GitHub the weights of the #FeNNixBio1 foundation machine learning model for drug design! 💫
Weights: https://t.co/QTLgyTZ3ad
FeNNol GPU code: https://t.co/ElYjDaNVDD
The models are distributed under the open source ASL license (i. e. restricted to non-commercial academic research).
You can also check the updated version of the preprint that includes a unified transformers architecture as well as the full computation of the Freesolv hydration free energies dataset etc...
https://t.co/eto2c2YXFO
Happy holidays and merry Christmas everyone! 🎅 🎄
Sorbonne Université / CNRS
@qubit_pharma
#machinelearning #moleculardynamics #drugdesign #compchem #GPU #biophysics
#compchem New preprint: "𝐀𝐜𝐜𝐞𝐥𝐞𝐫𝐚𝐭𝐢𝐧𝐠 𝐌𝐨𝐥𝐞𝐜𝐮𝐥𝐚𝐫 𝐃𝐲𝐧𝐚𝐦𝐢𝐜𝐬 𝐒𝐢𝐦𝐮𝐥𝐚𝐭𝐢𝐨𝐧𝐬 𝐰𝐢𝐭𝐡 𝐅𝐨𝐮𝐧𝐝𝐚𝐭𝐢𝐨𝐧 𝐍𝐞𝐮𝐫𝐚𝐥 𝐍𝐞𝐭𝐰𝐨𝐫𝐤 𝐌𝐨𝐝𝐞𝐥𝐬 𝐮𝐬𝐢𝐧𝐠 𝐌𝐮𝐥𝐭𝐢𝐩𝐥𝐞 𝐓𝐢𝐦𝐞-𝐒𝐭𝐞𝐩 𝐚𝐧𝐝 𝐃𝐢𝐬𝐭𝐢𝐥𝐥𝐚𝐭𝐢𝐨𝐧" in link with our #FeNNix-Bio1 foundation #machinelearning model.
👉 Check it out: https://t.co/iDXCzRZiH2
We present a strategy to accelerate molecular dynamics simulations using foundation neural network models. To do so, we apply a dual-level neural network multi-time-step (MTS) strategy where the target accurate potential is coupled to a simpler but faster model obtained via a distillation process. Consequently, large simulation speedups over standard 1 fs integration are observed: 4-fold in homogeneous systems and 2.3-fold in large solvated proteins. Such a strategy is applicable to any neural network potential and reduces their performance gap with classical force fields.
Great work from @comecattin. Thanks to all co-authors. Funding @ERC_Research (project EMC2) and supercomputing time @Genci_fr.
Sorbonne Université and CNRS
@qubit_pharma
#compchem Second preprint linked to the FeNNix-Bio1 #machinelearning foundation model. FeNNix-Bio1's inference is pretty fast already with a few GPUs but, "what if", we were able to push it at the #Exascale? Let's have a glimpse into the future (1/3):
"Pushing the Accuracy Limit of Foundation Neural Network Models with Quantum Monte Carlo Forces and Path Integrals"
Check it out 💫: https://t.co/4TsMVcuhQz
We propose an end-to-end integrated strategy to produce highly accurate quantum chemistry synthetic datasets (energies and forces) aimed at deriving Foundation Machine Learning models for molecular simulation. Starting from Density Functional Theory (DFT), a "Jacob's Ladder" approach leverages computationally-optimized layers of massively #GPU-accelerated software with increasing accuracy. Thanks to Exascale, this is the first time that the computationally intensive calculation of Quantum Monte Carlo forces (QMC), and the combination of multi-determinant QMC energies and forces with selected-Configuration Interaction wavefunctions, are computed at such scale at the complete basis-set limit. To bridge the gap between accurate QC and condensed-phase Molecular Dynamics, we leverage transfer learning to improve the DFT-based FeNNix-Bio1 foundation model.
🚀The resulting approach is coupled to path integrals adaptive sampling quantum dynamics to perform nanosecond reactive simulations at unprecedented accuracy on a full Satellite Tobacco Mosaic Virus (STMV) 1M, all-atom, complete solvated model (see the video produced using @VTX_mol, @MaximeMARIA_@MatthieuMontes ). These results demonstrate the promise of Exascale to deepen our understanding of the inner machinery of complex biosystems.
Immense thanks to all co-authors at @qubit_pharma, @LCT_UMR7616 (@Sorbonne_Univ_ /@CNRS ), @UChicago, @SandiaLabs, @ORNL and @argonne for this collaborative efforts. Some are on X: @QMC_Anouar@Thomas__Ple@OlivierApp@EPosenitskiy@blazhynska66497@ApplencourtT@JeongnimK #HPC
This work was made possible thanks to #INCITE projects enabling the use of Argonne's Aurora exascale system and of the Polaris machine, to @Genci_fr (Jean Zay @ Idris) and @EuroHPC_JU (Leonardo @Cineca1969). #supercomputing
#compchem What could we do if we had a fast and accurate foundation #MachineLearning learning model for condensed phase molecular dynamics simulation of biological systems?
👉Check the @ChemRxiv preprint: "A Foundation Model for Accurate Atomistic Simulations in Drug Design":
https://t.co/6KfB3EDN3A
Amazing work by @Thomas__Ple and the teams at @LCT_UMR7616 and @qubit_pharma introducing the FeNNix-Bio1 foundation model.
Trained exclusively on synthetic quantum chemistry data, it provides predictive condensed-
phase Molecular Dynamics simulations including quantum nuclear effects. Its full-range of capabilities is demonstrated by modelling: water properties, ions in solution, small molecules hydration free energies, complex folding free-energy landscapes, large-scale protein dynamics, protein-ligand binding, chemical reactions and by coupling it to protein structure prediction foundation models’ outputs for further refinement. FeNNix-Bio1 is accurate and systematically improvable while limiting human parametrization efforts.
Big kudos to all other co-authors: @OlivierApp, @QMC_Anouar, @EPosenitskiy, C. Villot, L. Lagardère. Implemented and #GPU-accelerated in Tinker-HP/Deep-HP @TINKERtoolsMD.
Funding @ERC_Research (project EMC2). A big thank to @Genci_fr (grand challenge H100 @ IDRIS/Jean Zay), @EuroHPC_JU (Cineca/Leonardo) and to @argonne (#INCITE project/Aurora) for #supercomputing time.
#compchem New preprint: Satellite Tobacco Mosaic Virus: Revealing Environmental Drivers of Capsid and Nucleocapsid Stability using High-Resolution Simulations.
https://t.co/LqvKAT9lLd
Large scale MD & metadynamics simulations of the complete STMV using the AMOEBA polarizable FF. Amazing work by @blazhynska. @Genci_fr@ERC_Research (projet EMC2).
https://t.co/LqvKAT9lLd
#compchem New group preprint: "Velocity Jumps for Molecular Dynamics".
https://t.co/PVkvcphQjL
We introduce the Velocity Jumps approach, denoted as JUMP, a new class of Molecular dynamics integrators, replacing the Langevin dynamics by a hybrid model combining a classical Langevin diffusion and a piecewise deterministic Markov process, where the expensive computation of long-range pairwise interactions is replaced by a resampling of the velocities at random times.
Fantastic job by @NicolaiGouraud and another nice interdisciplinary collab with Pierre Monmarché. @OlivierApp@Thomas__Ple@qubit_pharma@LCT_UMR7616@Genci_fr@TINKERtoolsMD
#compchem Just published in @ChemicalScience:
Water-Glycan Interactions Drive the SARS-CoV-2 Spike Dynamics: Insights into Glycan-Gate Control and Camouflage Mechanism. https://t.co/QkujjdHPQV
Towards developing therapeutic strategies against #COVID19, we performed μs-long all-atom AMOEBA high-resolution polarizable adaptive sampling molecular dynamics simulations and zoomed in on the #SARSCoV2 interaction layers in open and closed states. Simulations reveal a protein-solvent-glycan polarization network supporting the open state. Besides, we showed that the glycan shield maintains viral camouflage in both states. Amazing work by @blazhynska. Kudos to L. Lagardère and @OlivierApp. Another epic collaboration with @leucinw and @prenbme. This research was funded by @ERC_Research (project EMC2). We thank @awscloud for supporting this research and providing extensive #GPU-accelerated cloud computational ressources as well as @Genci_fr for #supercomputing time. #HPC
@LCT_UMR7616@qubit_pharma@Sorbonne_Univ_@CNRSchimie@UTBiomedical@TINKERtoolsMD
#compchem Happy to see this one out and published @JChemPhys as part of the special issue: "Modular and Interoperable Software for Chemical Physics":
"FeNNol: an Efficient and Flexible Library for Building Force-field-enhanced Neural Network Potentials.
FeNNol is a new #GPU-accelerated #opensource library for building, training and running force-field-enhanced #neuralnetwork potentials. It provides a flexible and modular system for building hybrid models, allowing to easily combine state-of-the-art embeddings with ML-parameterized physical interaction terms without the need for explicit programming. FeNNol is fast and intends to reduce the performance gap between ML potentials and standard (polarizable) force-fields.
JCP paper: https://t.co/0kahUQDW1l
Preprint (updated): https://t.co/Kyc3KnT7bO
Github: https://t.co/C9zaLhqa6P
Congrats @Thomas__Ple, @OlivierApp, L. Lagardère @LCT_UMR7616. Funding @ERC_Research (project EMC2). Supercomputer time @Genci_fr #supercomputing #HPC #machinelearning
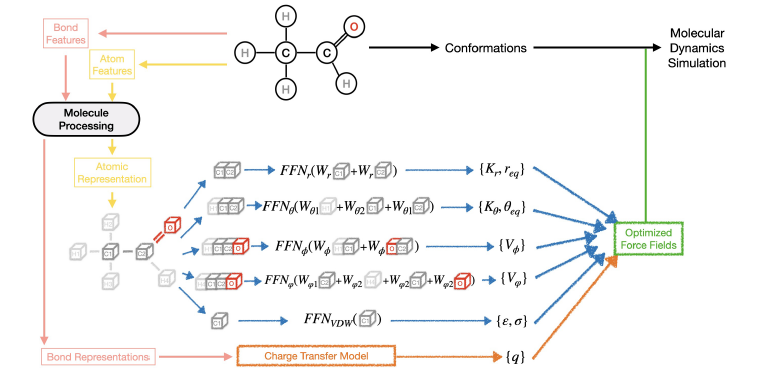
#compchem Just published in JCTC @JCIM_JCTC: "Advancing Force Fields Parameterization: A Directed Graph Attention Networks Approach".
https://t.co/a94pSRJj5y
In this study, we propose a Graph-Based Force Fields (GB-FFs) model to directly derive parameters for the Generalized Amber Force Field (GAFF) from chemical environments. Our end-to-end parameterization approach eliminates the need for expert-defined procedures & enhances the accuracy and transferability of GAFF across a broader range of molecular complexes. Nice work by G. Chen, @JaffrelotT, @Thomas__Ple, L. Lagardère & great collab with Y. Maday. Funded @ERC_Research (project EMC2). @Genci_fr
#compchem Check out this new paper where we reach chemical accuracy at the Full CI/CBS limit for small molecules (up to nearly 300 qubits!!) using GPU-accelerated quantum emulation.
#compchem Latest group preprint:
FeNNol: an Efficient and Flexible Library for Building Force-field-enhanced Neural Network Potentials.
https://t.co/Kyc3KnSzmg
A new GPU-accelerated #opensource library for building, training and running force-field-enhanced neural network potentials. It provides a flexible and modular system for building hybrid models, allowing to easily combine state-of-the-art embeddings with ML-parameterized physical interaction terms without the need for explicit programming. FeNNol shrinks the performance gap between ML potentials and standard force-fields.
Available at https://t.co/C9zaLhpChh
Great work by @Thomas__Ple, @OlivierApp, L. Lagardère @LCT_UMR7616.
Funding @ERC_Research (project EMC2).
Supercomputer time @Genci_fr. #NeuralNetworks #GPU #supercomputing #HPC
🥂As the end of year is nearly there, a quick recap on the 2023 @PiquemalGroup's papers & preprints in #MachineLearning for #compchem🤖:
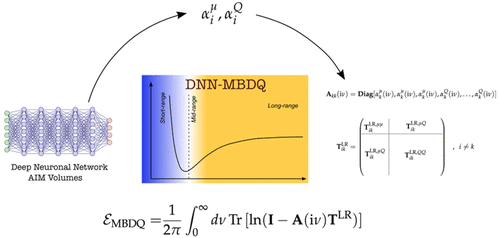
- Generalized Many-Body Dispersion Correction through Random-phase Approximation for Chemically Accurate Density Functional Theory; J. Phys. Chem. Letter @JPhysChem (Open Access) https://t.co/s5dRJ8W6h6
- Routine Molecular Dynamics Simulations Including Nuclear Quantum Effects: from Force Fields to Machine Learning Potentials; JCTC @JCIM_JCTC (Cover)
https://t.co/x3LVAl95m7
Preprint: https://t.co/0uFWLe3ZBS
- Scalable Hybrid Deep Neural Networks/Polarizable Potentials Biomolecular Simulations including long-range effects; @ChemicalScience (Open Access)
https://t.co/gpmY5ySnyX
- Force-Field-Enhanced Neural Network Interactions: from Local Equivariant Embedding to Atom-in-Molecule properties and long-range effects; @ChemicalScience (Open Access)
https://t.co/w77VOVps0M
- Incorporating Neural Networks into the AMOEBA Polarizable Force Field; preprint @ChemRxiv
https://t.co/cSJ0lsjOdh
- Advancing Force Fields Parameterization: A Directed Graph Attention Networks Approach; preprint @ChemRxiv
https://t.co/LHbHrW9mIg
Thanks to all our great co-authors: @Thomas__Ple, @JaffrelotT, @OlivierApp, @MaugerNastasia, @PierPoier, @olexandr, @leucinw, @prenbme, X. Wang, H. Gökcan, S. Huppert, Y. Maday, L. Lagardère.
@LCT_UMR7616. Funding @ERC_Research (project EMC2), supercomputer time @Genci_fr . This research uses the Tinker-HP @TINKERtoolsMD MD engine. Check it out at https://t.co/7bXXoamjO8
🚨Just published in @ChemicalScience🚨: "Force-Field-Enhanced Neural Network Interactions: from Local Equivariant Embedding to Atom-in-Molecule properties and long-range effects.
https://t.co/w77VOVps0M
Amazing #compchem work by @Thomas__Ple introducing the hybrid physically-driven #machinelearning FENNIX model. Exhibiting accurate gas-phase energy predictions, FENNIX is reactive, transferable to the condensed phase and able to produce stable Molecular Dynamics simulations including nuclear quantum effects. Funded @ERC_Research (project EMC2), supercomputer time @Genci_fr. @TINKERtoolsMD@LCT_UMR7616
#compchem Preprint:Lambda-ABF: Simplified, Accurate & Cost-effective Alchemical Free Energy Computations. New Colvars #opensource library for NAMD & Tinker-HP. Great efforts: L. Lagardère & L. Maurin & collabs: P. Monmarché & J. Hénin @IbpcF@LCT_UMR7616 https://t.co/Axei2msVfJ
@simonbatzner@jppiquem There are F atoms in molecules of the DES370K dimers. Though we only train on the dimer interaction energy and not on individual monomers. So the H-F covalent stretching would be a very indirect information.