AI for novel biosensor design. I asked Claude to test SwitchCraft to design a potential binder for an immune signaling molecule, cGAMP.
In our cells, we have the STING protein, which initiates an immune signaling cascade when it binds cGAMP.
To design a potential binder of cGAMP, Claude loaded up SwitchCraft on a neocloud (Lambda) and generated several designs: small proteins, unrelated to STING, predicted to change shape when cGAMP binds.
Claude then used PyMOL and ChimeraX to create this presentation on the design.
Binder design has come of age thanks to generative models—but how can we access the wider array of dynamic, multistate protein functions, so elegantly employed by nature?
@mihirbafna14 and I are excited to share SwitchCraft, a framework for designing such functions. (1/7)
Here's an ESMFold2 demo run by AI agents on Modal for designing potential GLP-1 receptor binders.
This test focused on the GLP-1R extracellular domain where semaglutide/Ozempic binds. It measured how well each design recovered the residues that semaglutide contacts.
Codex made this neat presentation of the demo with autonomous use of ChimeraX and ElevenLabs.
I’m so excited about the launch of ESMFold2, ESMC, and the new ESM Atlas. This was a massive team effort, and I’m grateful to have worked with such an incredible group @biohub.
A headline result I’m especially excited about: ESMFold2 can design minibinders and antibodies with nanomolar affinity, target selectivity, and functional activity against therapeutically relevant targets.
Today, we’re sharing the full binder design protocol.
Grok Build with Composer 2.5 visualizing predicted binders against PD-L1, a cancer immune-checkpoint protein that tumors use to switch off T-cells.
Composer 2.5 in Grok Build is using ChimeraX to visualize PD-L1 binders generated by Genie 3. In this workflow, a region on PD-L1 was selected for binding, Genie 3 generated binder shapes, ProteinMPNN filled in the amino acid sequences, and Boltz folded the binders against PD-L1.
We are excited for all of you to try out Composer 2.5 in Grok Build starting today!
To use composer-2-5 do `/model` in Grok Build and type in Composer to switch
Composer 2.5 comes with 200k context window and supports: subagents, MCPs, skills and additionally also works with your .cursor settings
For researchers interested in using AI agents in their bioinformatic workflows related to genome mining and gene discovery, you can point your preferred agent (e.g. Claude Code, Codex) at this skill repo I put together.
This repo provides an additional harness layer for structured gene cluster analysis, bioinformatic tool use, and comparative pathway mapping. If you are 'compute poor' like many researchers, this repo helps your agent use cloud/neocloud resources such as AWS, Runpod, and Lambda.
It also nudges your AI agent to keep logs and learnings so your agent can adapt and get better at your workflows.
https://t.co/fZdMZaEKWJ
AI for genome mining.
Many high-value natural compounds are made by enzymes whose genes group into clusters. For many compounds the pathway is not fully elucidated. Finding those genes, and later testing them, helps piece a pathway together.
For instance, a gene that is near a known anchor gene becomes a candidate for a missing step, even when the anchor is only an approximate match.
As a demo, Claude picked berberine as a target compound, then ran bioinformatic tools across different plant lineages to compare the pathway's gene architecture and flag candidate genes for open steps.
Claude also made this short presentation!
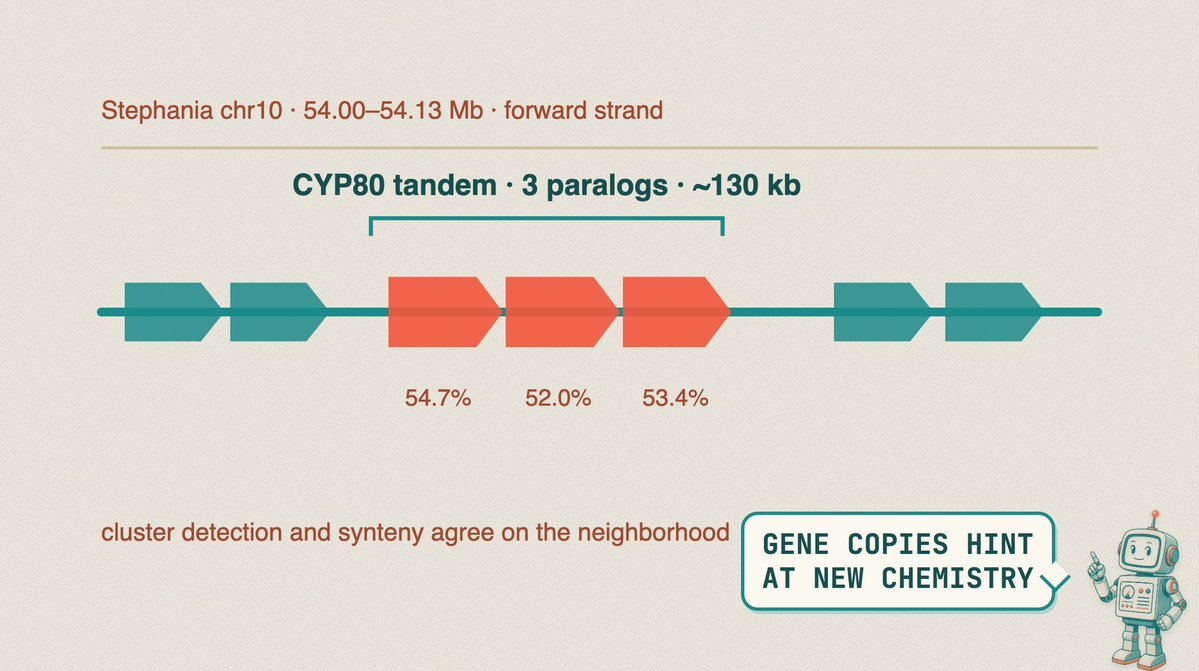
Claude searched four plants for gene clusters related to the berberine pathway. This included the berberine bridge enzyme (BBE), the CYP80 and CYP719 P450s, and O-methyltransferases.
1. Chinese goldthread (Coptis chinensis): the canonical berberine/BIA pathway reference
2. Fourstamen stephania (Stephania tetrandra; TCM han fang ji): known for coupled bisbenzylisoquinoline alkaloids like tetrandrine
3. Fish mint (Houttuynia cordata): where Claude found many BBE-like paralogs spread across the genome
4. Amur cork tree (Phellodendron amurense): a published P450 route, used as comparative context
Here is an example where Claude found a tandem of three CYP80-like paralogs beside an anchor gene.
@arjbalaji Thanks! Works well with CC and codex. Should also work well with other harnesses which can handle multiple tool calling, compaction, and monitoring loops.
AI for structural biology: given the right harness, tools, and compute, your preferred AI agent can tackle tasks such as building a protein structure from a cryo-EM density map.
In this case, Claude assembled a structure, compared its reconstruction to the published structure, then used ChimeraX to create these visuals and make this short presentation video.
For structural biologists comfortable with tools like Claude Code or Codex, you can point your AI agent at this skill repo to give your agent a better harness for cryo-EM and related structural workflows.
Also, a big issue with a lot of structural work is the amount of compute and storage needed. Neoclouds are great for this, and this repo points your agent at some of these resources. It also nudges your agent to set up self-learning / memory so your agentic workflows can improve over time.
https://t.co/7IJ7rxfYgZ
Claude also compared several tools for building the protein from the cryo-EM density. In this case, Claude used ModelAngelo, CryoAtom, and Cryo2Struct, and then constructed this dashboard to display the results.
For those interested in trying out ESMFold2, you can use the Biohub API or run the Hugging Face weights directly.
Here's an example of codex using ESMFold2-Fast weights on a neocloud (lambda ai) for predicting the structure of a small VIPR protein + RNA complex. Codex used ChimeraX for the visuals.
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
This is what it looks like to have your AI agent run your favorite structural biology tool. In this case, codex ran the new ESMFold2 on the cloud for a small VIPR RNP and then autonomously created snapshots of the results in ChimeraX.