🎉 Thrilled to share that my first-author paper as a PhD student is officially in Cancer Discovery now:
https://t.co/W7oEwrry4e
Huge thanks to my mentors for their invaluable guidance, and to all my amazing collaborators for their support and insights throughout this journey.
Our recent work on COVID-19 single cell meta-analysis using ToppCell.
https://t.co/jkqJtc1mGC
Welcome to play with the data on ToppCell COVID-19 signature atlas on https://t.co/0E0V3W2IkL
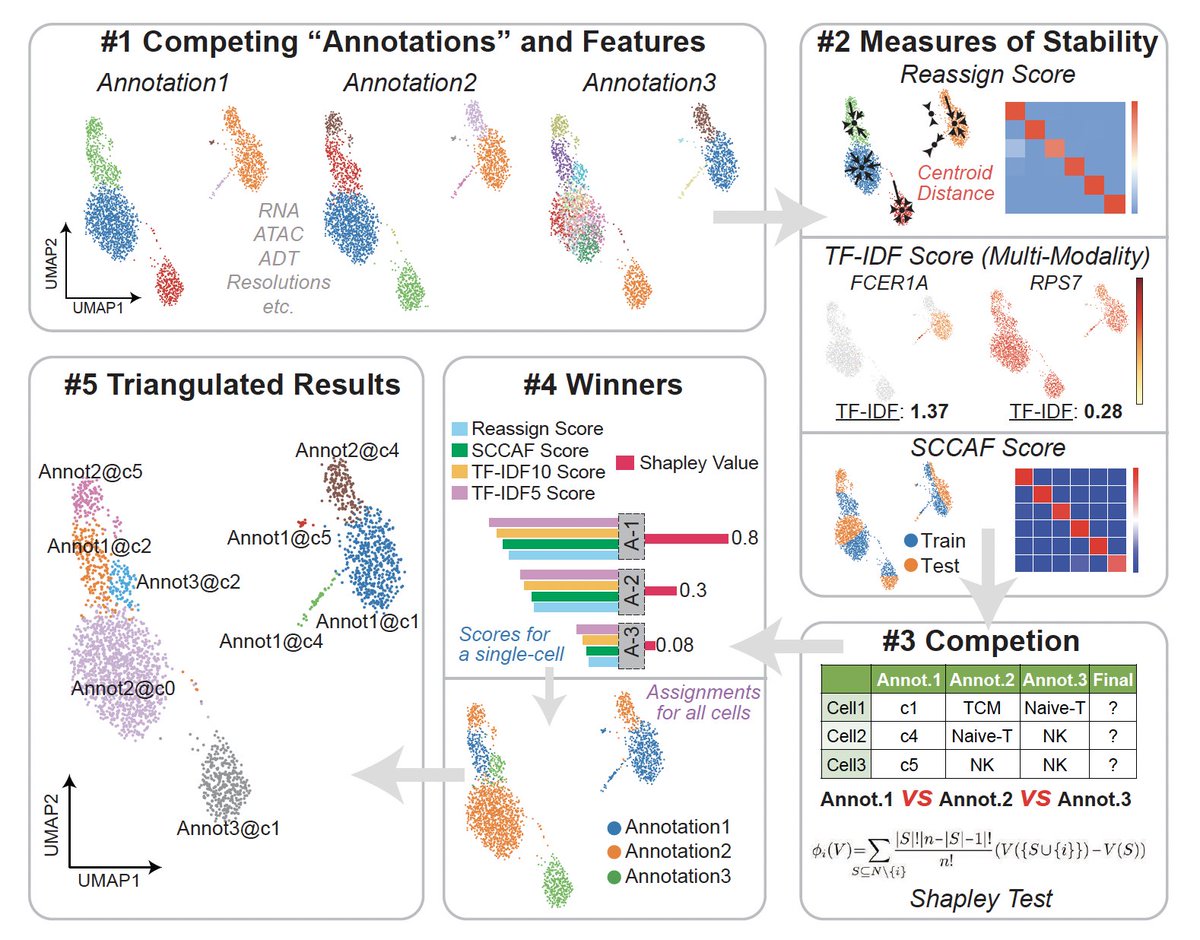
Happy to share our latest computational tool -- scTriangulate, leveraging cooperative game theory to mix-and-match diverse cluster boundaries to achieve the optimal clustering results! (1/n)https://t.co/Z2iSWloeuT
ATAC peak calling with MACS2: I know this is a recurring problem but I got asked a few times recently, so I just put all info here for my own ref. We now routinely convert paired BAM to simple BED file and use "-f BED --shift -100 --extsize 200" for the peak calling. Why? 1/7
@tangming2005 Have you checked out https://t.co/WQK0x2Hpwy that's a cool resource offered by embl provides link outs to publications using the tool and altmetrics.
there are so many genome fasta and gtf files available from different resources: UCSC, Ensembl, genecode. which files are recommended for RNAseq and DNAseq alignment? Heng's post answered some of it https://t.co/IivSogOhch what about gtf, gtf should be compatible with fasta