Excited to share our #RECOMB2026 work, DETANGO: a deep learning framework for disentangling mutation effects on protein stability and function from evolutionary signals captured by protein language models (pLMs).
DETANGO estimates a functional plausibility score that quantifies mutation effects on function beyond what can be explained by stability alone. Across extensive benchmarks, DETANGO accurately identifies stable-but-inactive (SBI) variants and functionally important residues involved in ligand binding, catalysis, and allostery.
Grateful to my co-authors @ZiangLi2001, @Qwe1029384756Tu, and Jiaqi Luo, and to my advisor @luoyunan for their invaluable contributions and support throughout this work! Special thanks to Tony for representing our team and presenting DETANGO today at RECOMB 2026 in Thessaloniki, Greece!
Preprint: https://t.co/xwLTYSRFM6
Code: https://t.co/HqnrlI56Na
#MutationEffectPrediction #ProteinLanguageModels #ProteinFunction #ComputationalBiology #DeepLearning
Excited to share that SPURS is now published in Nature Communications! 🎉
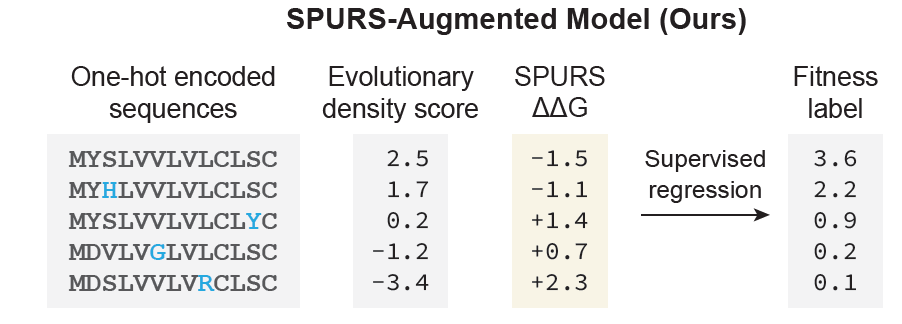
SPURS is a fast, accurate, and generalizable protein stability predictor that enables learning about protein function through the lens of stability. This work was done with my wonderful advisor @luoyunan!
@luoyunan We apply SPURS to examine the role of protein stability in pathogenicity, revealing that stability loss contributes to pathogenicity in different ways across proteins. 10/n
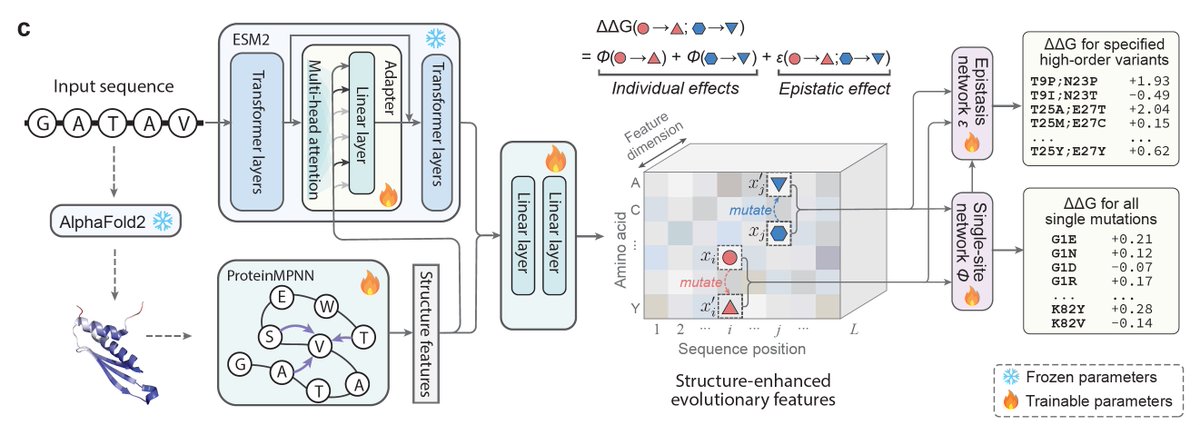
@luoyunan Given AlphaFold2-predicted structures and sequence, SPURS can be combined with ESM1v to identify functional sites in an **unsupervised** manner. 7/n
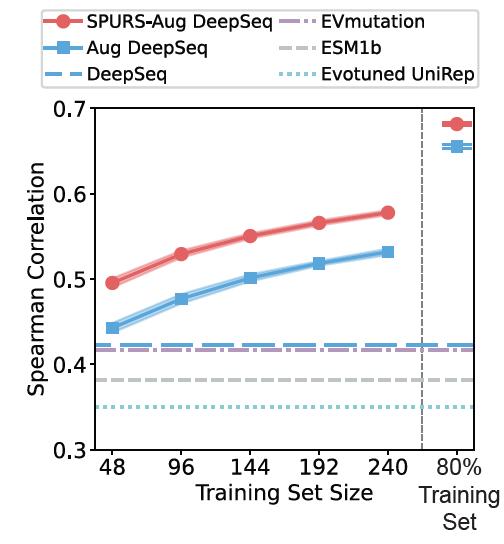
@luoyunan We find that SPURS-predicted stability serves as a natural prior for protein function, since a protein must remain at least marginally stable to function. By combining sequence, structure, and predicted stability, we explain functional variation across 3 downstream tasks. 6/n
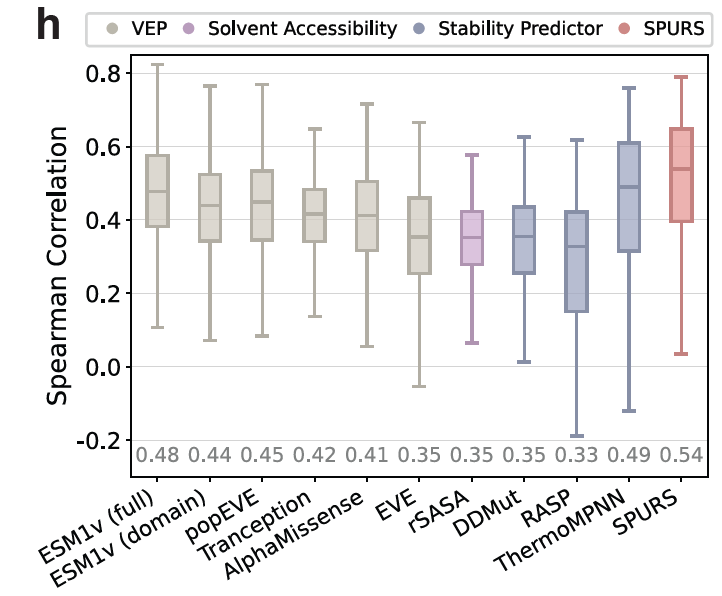
@luoyunan Compared to traditional biophysics-based tools, Foldx and Rosetta, SPURS is substantially faster and achieves higher accuracy in our evaluations. 5/n
@luoyunan Inference with SPURS scales as O(1) (one forward pass) per protein, enabling site-saturation mutagenesis of the entire human proteome of ~19,652 proteins (~10⁹ variants) in 30 minutes on a single GPU. 4/n
@luoyunan SPURS performs strongly on Domainome, a recently released large human domain site-saturation mutagenesis dataset of 500 human protein domains, exceeding previously reported results. 3/n
@luoyunan SPURS achieves state-of-the-art performance across 11 benchmarks and generalizes across stability tasks including ΔΔG optimization and protein melting temperature prediction. 2/n