He estado en el orgullo de Madrid y he podido ver a muchas de mis DRAGS favoritas.
Decir que han sido todas un amor y me ha hecho mu feliz hablar con ellas.
🧵Hilo del contenido audiovisual:
No scaling laws for single-cell foundation models: when bigger atlases stop teaching the model anything
In language and vision, the recipe has been simple: more data, bigger models, better performance. Single-cell biology borrowed that playbook. Foundation models for transcriptomics jumped from 1 million cells to atlases of over 100 million, on the assumption that scale would unlock the same gains. Alan DenAdel and coauthors put that assumption to the test, and the result is sobering.
Working from a 22.2-million-cell corpus, they pretrained 400 models across five architectures (from PCA and a variational autoencoder up to the Geneformer transformer) and ran 6,400 evaluation experiments. They varied not just dataset size (1% to 75%) but also diversity, using cell-type re-weighting and geometric sketching to deliberately enrich rare cell types and transcriptional states.
The finding: performance saturates almost immediately. On cell-type classification, batch integration, and perturbation prediction, most models hit their ceiling at roughly 1% of the corpus, about 200,000 cells. Beyond that, adding millions more cells changed essentially nothing. More diversity didn't help. Even spiking in genome-scale Perturb-seq data, to give the models perturbed phenotypes rather than just healthy ones, failed to move the needle. Larger models did score better overall, but they too plateaued early on data.
Two points stood out. Simple baselines (PCA, logistic regression) often matched or beat the transformers. And the strongest model, SCimilarity, won not because of size but because its contrastive training objective is aligned with the downstream task. For single-cell data, what you train on and how you frame the objective matters far more than how much you collect.
This reframes a quiet but expensive habit. In drug discovery, biotech, and any pipeline leaning on cell atlases, the instinct to keep scaling pretraining corpora may be burning compute for no return. The real leverage sits elsewhere: curating high-quality, task-relevant data and matching the training objective to the actual question you're trying to answer.
Paper: DenAdel et al., journal license | https://t.co/X7GxoxF5U5
Gracias, Presidente, por este reconocimiento, por defender una Europa que pertenezca a sus pueblos, en el respeto del derecho internacional, no por encima ni en contra de él, y por la solidaridad, linfa vital de la familia humana.
Palestina libre, y nosotros con ella.
Neural networks might speak English, but they think in shapes.
Understanding their rich *neural geometry* is key to understanding how they work – and to debugging and controlling them with precision.
Starting today, we’re releasing a series of posts on this research agenda. 🧵
🇺🇲 Trump " İtalya bu Zorlu zamanda bizim yanımızda durmadı "
🇮🇹 Giorgia Meloni " Ben bir kadınım ve Aynı zamanda bir anneyim. Masum Çocukların katilinin yanında duramam "
Title:
Global genetic interaction network of a human cell
DOI: 10.1016/j.cell.2026.00345-4
A major bottleneck in human biology has been the inability to systematically map functional relationships between genes at scale. In this new Cell study, researchers construct a global genetic interaction network in human cells, providing one of the most comprehensive frameworks yet for decoding cellular function.
Using large-scale perturbation strategies, the study maps gene co-essentiality and genetic interactions across the genome, revealing how genes cooperate, buffer, or compensate for each other under physiological conditions. �
Cell
A key conceptual advance is the transition from single-gene essentiality to network-level functional architecture. Rather than asking “is this gene essential?”, the work asks:
👉 what genetic context makes a gene essential?
This enables identification of:
• Functional modules (protein complexes, pathways)
• Redundant buffering systems (synthetic rescue)
• Vulnerabilities (synthetic lethality candidates)
Importantly, the network reveals conserved interaction principles across evolution, suggesting that cellular organization follows reproducible design rules—not random complexity. �
Cell
From a translational perspective, this is highly actionable:
• Cancer therapy: pinpoint synthetic lethal targets in tumor-specific contexts
• Precision medicine: interpret variants through network position, not just mutation
• Drug discovery: prioritize pathway-level interventions over single targets
Conceptually, this work shifts biology toward a systems-level causality model, where phenotype emerges from interacting gene circuits, not isolated components.
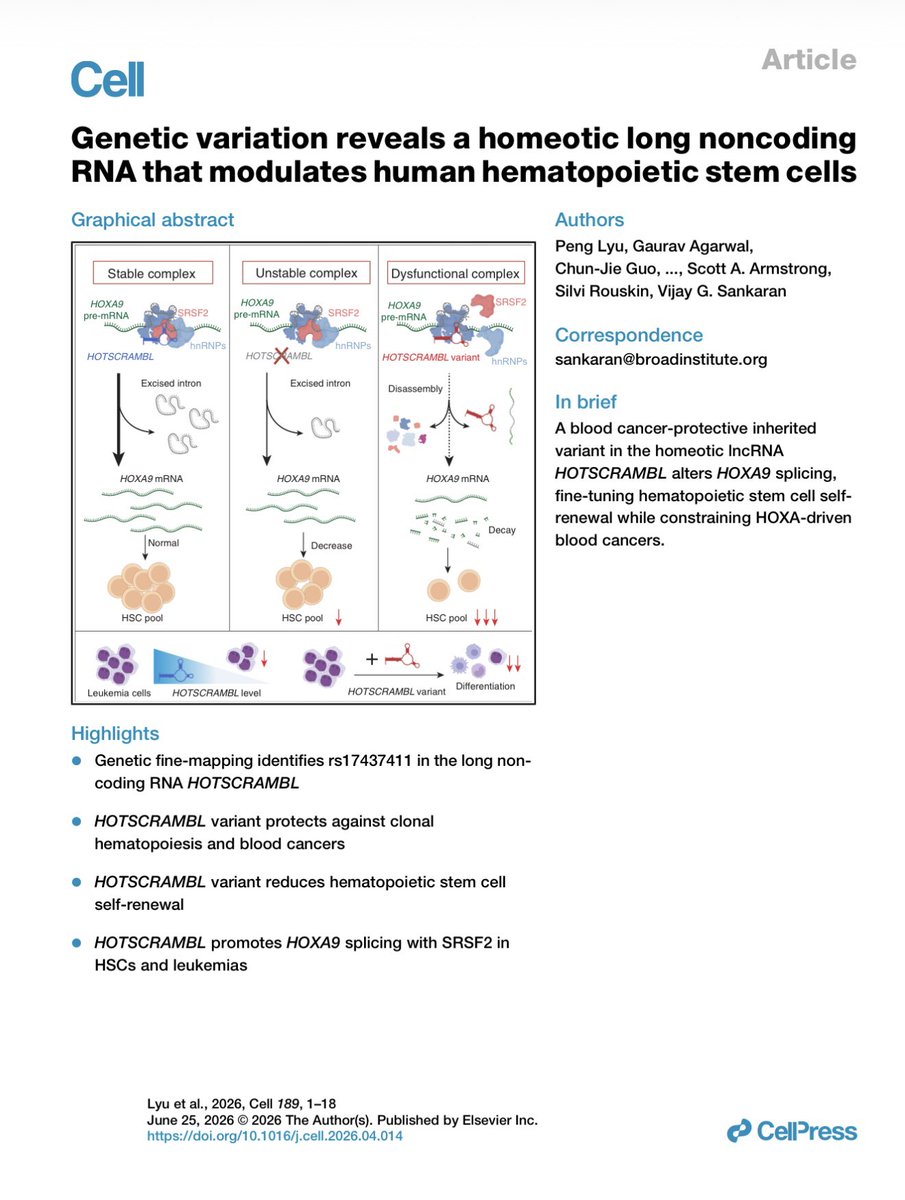
Check out super cool new paper we contributed to , I’m calling it “"One SNP refolds a lncRNA and rewrites the rules of stem cell self-renewal.” A new lncRNA called HOTSCRAMBL sits in the HOXA cluster and works as a splicing chaperone — it recruits SRSF2 to help HOXA9 get properly spliced in blood stem cells. A common variant (rs17437411) doesn't destroy the RNA, it refolds it, hiding the SRSF2 binding sites. The result: a dysfunctional decoy that can't scaffold splicing, HOXA9 levels drop, stem cells lose self-renewal, and leukemia growth gets constrained. The variant hits harder than a full knockout because the misfolded RNA actively interferes. It's essentially a built-in genetic brake on blood cancer. Read more: https://t.co/cnmeg0r015
Sí existía lo LGTBI.
Y sí, fuisteis referentes.
No por vosotros, sino por lo que significabais para la gente entonces.
Que ahora no lo seáis es otra historia.