Super excited to share my lab's latest work on quantifying the private information leakage from single cell count matrices. Led by very first postdoc in the g2lab, brilliant @BioConorWalker, we showed that individuals can be reidentified 1/n https://t.co/q9R60SvdV3
If you were looking for evidence that isoforms are cool, all-pervasive in the 🧠, and everybody should study them at the single cell level, our work on mouse development and brain-region variability from the @hagentilgner lab is now live @NatureNeuro! 1/
https://t.co/VUJlSu979C
A few months ago we posted a pre-print regarding a new approach to likelihood-based phylogenetics at short divergence scales (e.g. genomic epidemiology). I have now further optimized the code and updated the preprint https://t.co/K8chTGu9cS
This is a brilliant preprint. It describes an experimental method for separately sequencing the paternal and maternal genomes. This goes beyond the standard "experimental phasing" - the method can also tell which chr is maternal and which is paternal.
https://t.co/zZpHVLrvr2
We've known for a long time that a large fraction of noncoding GWAS hits are not currently explained by eQTLs. Why?? I'm excited to share our new preprint on this! Take home: GWAS and eQTL assays maximize power for different types of variants. (1/n)
https://t.co/JUFXnE7zLU
Groundbreaking work from @Nicola_De_Maio@EBIgoldman and colleagues
"we rewrite the classical Felsenstein pruning algorithm so that we can infer phylogenetic trees on at least 10 times larger datasets with higher accuracy than existing maximum likelihood methods. "
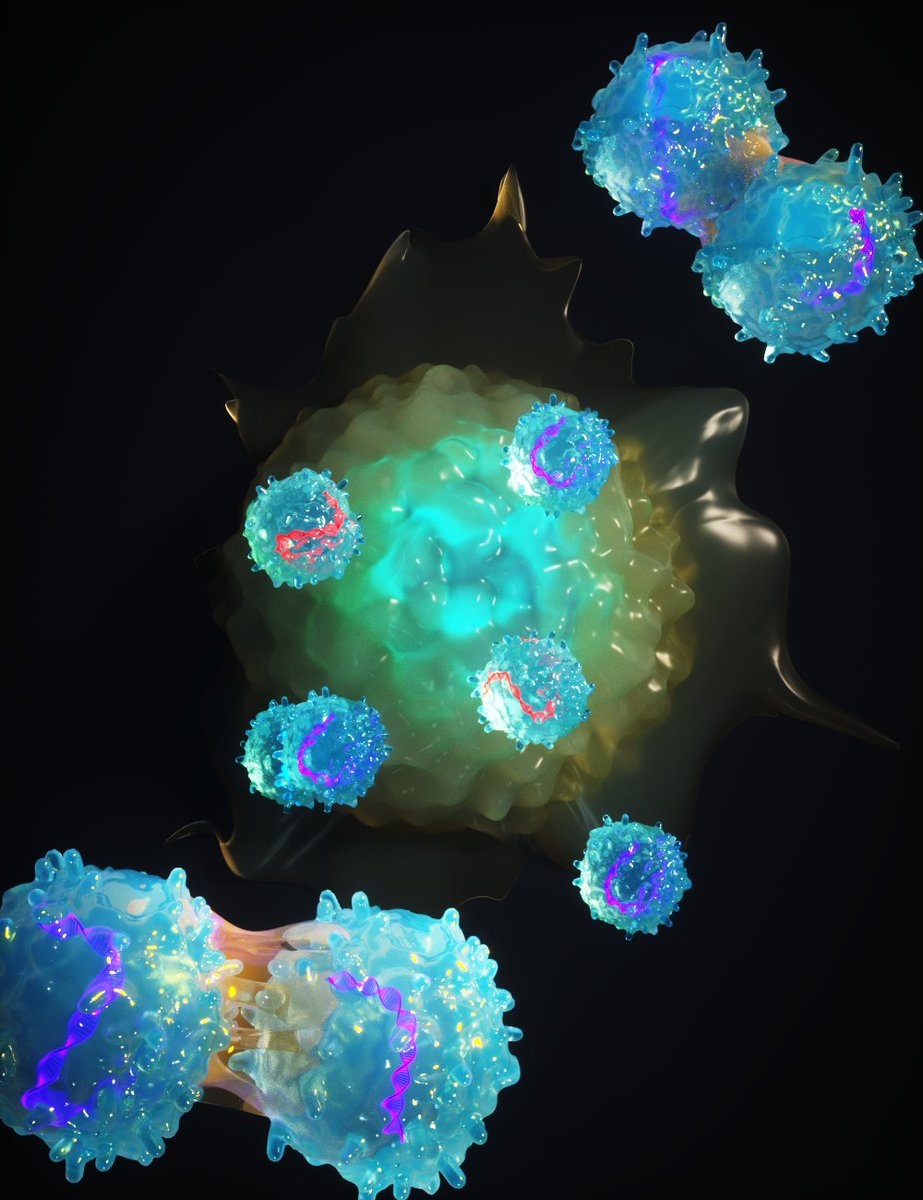
Really chuffed to share our latest work from Sanjana Lab out in @nature today (https://t.co/skHTGrKACw).
We tested >12,000 genes to find positive regulators of T cell proliferation to be used for next-gen #immunotherapies.
A thread...
Looking forward to starting my new position with @gamzeandgursoy at @nygenome! I will be working on developing methods for genomic #privacy.
https://t.co/vPJQxSCDNT
Simulating pandemic-scale genome sequencing datasets, e.g to benchmark your new analysis methods? Try phastSim, now updated to include indels (https://t.co/4V9xAf2qEm)
[1/2]
Glad to see this out. Here, we track ~700 clones across >300 individuals over ~13 years to unravel key aspects of clonal haematopoiesis (when there are genetically-defined subpopulations of haematopoietic stem cells in your bone marrow).
We are still looking to hire a postdoc bioinformatician on this fascinating project. Please read/RT/pass on to anyone who might be interested:
https://t.co/dkTNhOfVdF
We are re-advertising this position! Please check-out 👇 the original thread 👇, pass to anyone you think might be interested, share with colleagues, RT ...
https://t.co/02VoSI3GAO
The impressively huge scale of SARS-CoV-2 sequencing has really stretched our ability to do phylogenetics, and is leading to some fantastic advances in methods. Here's one that @Nicola_De_Maio from the @EBIgoldman group at @emblebi contributed to.
https://t.co/SlqtnTsD1H
Postdoc position in @EBIgoldman Group at @emblebi, to join new multinatl collab project creating proof-of-principle high-throughput nanopore device for determining amino acid sequence and identity of proteins
Full details at https://t.co/2FTwXNrn6G
<1/2>
For those of you who love to explain complex mutation clusters parsimoniously (who doesn't?), the final version of @BioConorWalker's paper is now out at https://t.co/aRHLjXKkh4 - also feat. @aylwyn_scally and @Nicola_De_Maio. And me. Cool figures too: this 👇 is part of S.Fig 10



























![EBIgoldman's tweet photo. Simulating pandemic-scale genome sequencing datasets, e.g to benchmark your new analysis methods? Try phastSim, now updated to include indels (https://t.co/4V9xAf2qEm)
[1/2] https://t.co/IfLzHZ1wok](https://pbs.twimg.com/media/FADWocgXoAIStrE.jpg)






















