Thank you to the all people who showed interest in my work and to all the others who attended and organized this year's @LatinXChem event - it was, as always, a pleasure! 😊😌✨️ #LatinXChem23 (1/2)
@icorraluam Adding the vibrational contribution to the broken symmetry approach unfortunately still yields comparatively poor results since the important reshift that arises from the Spin-Flip treatment is missing (2/2)
@icorraluam Hi Ines, sorry for the late reply! We did not apply any shifts to the computed spectra, the observed shifts arise purely from the methods used in the computational protocol (1/2)
@BoadoCercis@LatinXChem@digital_rsc@PCCP@ChemRxiv@LufacIDT Hi Cercis, we used CAM-B3LYP for all TD-DFT calculations, I did not put it on the poster, that's my bad :)
We unfortunately did not (yet) explore the potential for conical intersections, we are only just now beginning to understand the excited states of the system
@GustavMondragon@LatinXChem@digital_rsc@PCCP@ChemRxiv@LufacIDT you observe a redshift in the singlet manifold, with the band previously at around 400 nm shifted to around 550 nm and an additional band “pushed out” of the manifold to around 400 nm which leads to better agreement with the experimental result (3/3)
@GustavMondragon@LatinXChem@digital_rsc@PCCP@ChemRxiv@LufacIDT and variations thereof but very quickly ran into the same problem of computational feasibility. Luckily, Spin-Flip TD-DFT retains some amount of non-dynamical correlation by design, even in a DFT framework, which is why compared to the standard broken symmetry approach, (2/3)
And 3) Simulating vibrational motion via quantum sampling
If you'd like to know more about these particular approaches, or if you have any other questions, don't hesitate to ask! (4/4)
1) Increasing electronic flexibility by enabling access to more single-excitation configurations through a Spin-Flip TD-DFT approach
2) Including a solvent environment and ensuring correct description of long-range interaction (3/4)
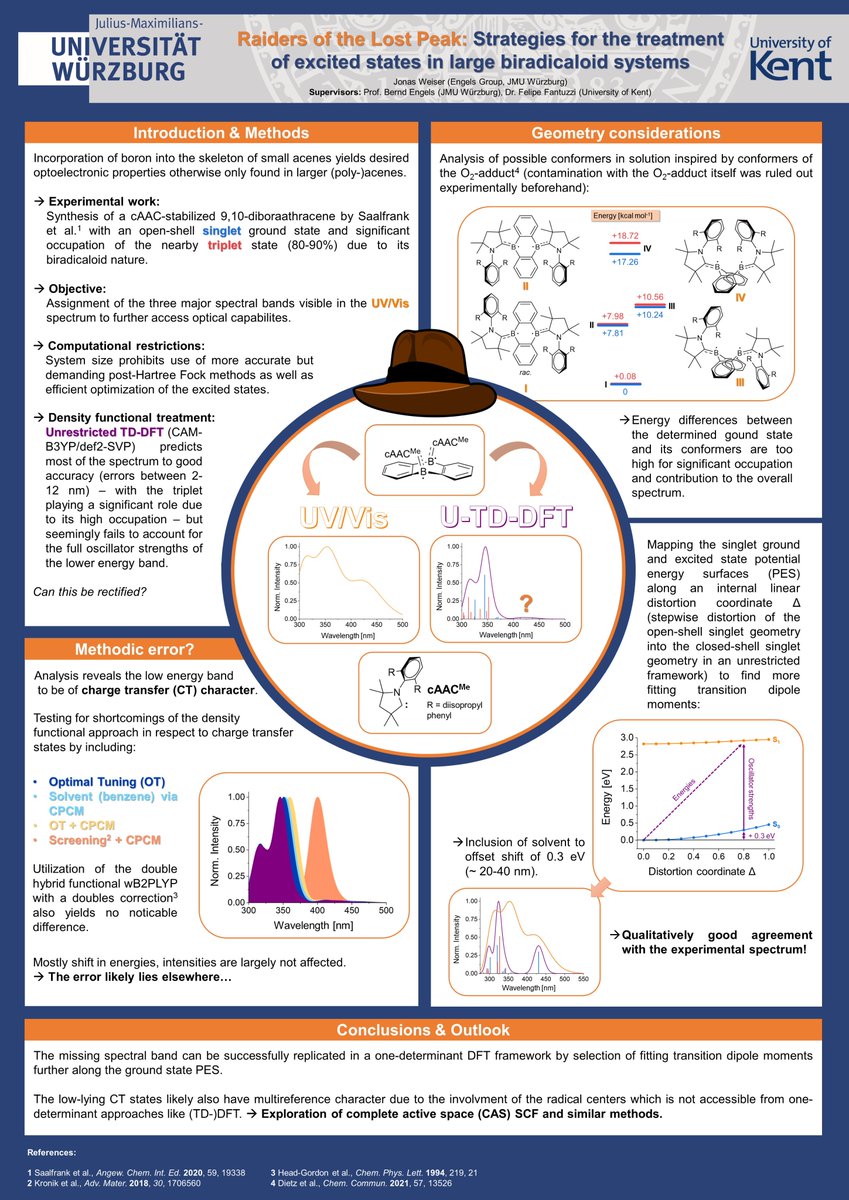
Just accepted in @J_A_C_S: Maximilian Dietz's (@maxxdietz) latest work on structure and electronics of CAAC-stabilized diboron-doped acenes! #chemtwitter#boron#maingroup
https://t.co/raVpww7wsh
@rubivf@LatinXChem since that program suite is, generally speaking, more robust.
Concerning the family of Truhlar functionals: I have not tried them yet, but it is on my to do list (especially because of their performance for CT states) :) (2/2)
'Excited' to present part of my work on the Excited States of Carbene-Stabilized Diboraacenes to the @LatinXChem community, it has been an adventure!🤠
Feel free to like, retweet, share and most importantly, discuss!😁
#LatinXChem22#LXChemComp#Comp091#DFT#boron#ExcitedStates
@rubivf@LatinXChem Hi Rubicelia, thanks for your questions! I'm mainly using Orca for the TD-DFT calculations due to the efficient SHARK integral engine of its most recent 5th version, though preliminary geometry optmizations have been done in Gaussian 16 (1/2)
Hi @LatinXChem, I am Felipe Fantuzzi 🇧🇷, an Assistant Professor (or Lecturer in Chemistry using 🇬🇧 titles) at @UniKent. Today I will introduce you to some of the great students/researchers that I have the privilege of mentoring and their #LatinXChem22 posters! Follow the 🧵!
Hi @LatinXChem, I am excited to share my work ‘Mechanistic insights into the collision-induced structural transition of graphene nanoflakes into carbon nanotubes’ with you! #LatinXChem22#LXChemComp#Comp102
Hi @LatinXChem, this is my work 'A Theoretical Investigation Of Astrophysically Relevant MgCnH Magnesium-Bearing Carbon Chains And Related Isomers' #LatinXChem22#LXChemComp#Comp006

























![JhoanLondoo4's tweet photo. Hi @LatinXChem, I'm Jhoan and this is my work “X-Ray & UV Photoinduced Fragmentation of Prebiotic Molecules in Interstellar Clouds: Ethanolamine”. For more information, just follow the thread! [1/10]#LatinXChem22 #LXChemPhys #Phys37 #Astrochemistry #GasPhase #Synchrotron https://t.co/bctGQTPGhS](https://pbs.twimg.com/media/FipiUzKXwAAvkcX.jpg)






















