Pakistan Genomic Resource (PGR), founded by Danish Saleheen, is the world's largest genetic database of human knockouts. It has been quietly building since 2017, from 10,000 individuals (Saleheen et al. Nature 2017) to nearly 200,000 today.
A new paper in Nature by Koch et al. reports a new analysis of 173,303 Pakistanis from PGR, offering genetic insights from natural inbreeding experiments in humans.
Koch et al. Nature 2026
https://t.co/Uk9YaeN1Ev
In biology, to understand what a gene does, you delete it, a standard experiment. Knock out the gene in a mouse and see what breaks: organ fails, behaviour changes, animal dies. Thirty years of biology built this way. It works, until it doesn't. Because, you know, mice are not humans.
To know the function of a gene in humans, you need a similar experiment. But that's unethical, you cannot deliberately delete a gene in a human. Except you don't have to, because nature has already been doing that experiment.
Occasionally, a person inherits a broken copy of a gene from both parents and is born with no working version at all--a human knockout. The problem is finding them. In most of the world's populations, where mating is largely random, these events are nearly invisible. For a gene-inactivating variant at 0.1% frequency, you'd expect one homozygous individual in every million people.
That math collapses in populations like Pakistan, where consanguineous marriage has been practised for centuries. In first-cousin marriages, the odds of observing a human knockout for the same 0.1% variant rise to roughly 1 in 16,000, a 63-fold enrichment. And the rarer the variant, the larger the advantage: ~630-fold for a 0.01% variant, ~6,300-fold for a 0.001% variant. The variants too rare to ever be seen in European biobanks become findable here.
Sequencing more than 170,000 individuals from highly consanguineous communities, the authors report a mind blowing statistic: at least one living human knockout was observed for 6,476 genes, which is nearly 1/3rd of the entire protein-coding genome!
What do we find when we finally have the human knockouts?
Studying the phenotypes in the human knockouts helps us confirm or refute our understanding of the gene's function based on animal studies. A few examples I highlight below.
PRDM9
PRDM9 might be one of the most popular genes among animal biologists. It encodes a protein that controls where chromosomes break and recombine during sperm and egg formation. Deleting the gene has caused infertility in every animal. PRDM9 was classified as the first hybrid sterility gene in vertebrates, so fundamental that crosses between mouse species with different PRDM9 alleles can't produce a fertile offspring. PGR now has 4 human PRDM9 knockouts : three women, one man. All fertile, with 2 to 7 children each. A 14-year biological fact, overturned by four families in Pakistan.
LRRK2
LRRK2 is a well-established Parkinson's disease risk gene. Activating mutations in LRRK2 are among the most common risk factors for Parkinson's. LRRK2 is a therapeutic target with many companies exploring ways to switch off this gene in the brain to treat Parkinson's. Large-scale sequencing studies have found individuals with partial loss of LRRK2, who did not show any concerning health issues, predicting adverse effects of LRRK2 inhibition in humans. Animal knockouts though warned of kidney damage. Now PGR has two LRRK2 knockouts, both with kidney disease, confirming animal studies.
RXFP1
RXFP1 encodes the receptor for a pregnancy hormone called relaxin, which has long been studied in rodents. Animal studies suggested it played a critical role in cardiovascular adaptation and connective tissue remodelling, fuelling relaxin-targeted drug development, which failed in late-stage trials. PGR found 16 RXFP1 knockouts, expanded to 26 via recall-by-genotype, all tested with cardiac imaging. None had consistent cardiovascular or reproductive deficits, retrospectively explaining the failure of relaxin-targeted drug programmes that might have spent millions of dollars. Mouse physiology failed to inform humans in the case of relaxin.
Gene constraint insights
Existing large-scale biobanks are predominantly European-based outbred populations, which shaped our understanding of gene constraints largely based on intolerance to partial loss of function. Now PGR, a South Asian-based cohort enriched for consanguineous communities, is beginning to offer insights into gene constraints based on intolerance to complete loss of function.
PGR showed that nearly 1/3rd of human genes tolerate complete loss of function. As much as the genes for which knockouts were found, the genes for which knockouts weren't found can offer biological insights.
The authors find genes depleted for knockouts in PGR are enriched for genes essential for cell survival, known Mendelian disease genes (both dominant and recessive) and genes broadly expressed across human tissues.
A fascinating insight is significant enrichment for knockouts in tissue-specific genes (OR=2.39). Human knockouts confirm what drug developers have always thought: tissue-specific genes are much safer therapeutic targets than broadly expressed genes.
The above insight should be read with caveats. The sample size of PGR is small, hence not saturated for human knockouts. It's likely the number of genes will increase as the sample size grows. There is a survivorship bias, like any other volunteer-based cohort. Absence of a gene knockout here doesn't mean biological impossibility. It means incompatibility with being a 'healthy' adult volunteer. If you build a cohort based on a hospital-based pediatric rare disease South Asian cohort, you'd expect to see knockouts that never appeared in PGR.
South Asian populations represent nearly a quarter of humanity, yet they have been largely absent from the genomic revolution. PGR shows what absence has been costing the field: overturned biological assumptions, failed trials, missed targets. The biology was always there. We just weren't looking in the right place.
Sorry to all the folks at Meta who lost their jobs. Tough times. Claiming AI will result in an amazing job boom or the opposite that they will completely destroys all jobs are both extreme positions. 1/
David Reich is back.
He and collaborator Ali Akbari just published a paper that overturns a long-standing consensus about human evolution — that natural selection has been dormant in our species since the agricultural revolution.
By scaling ancient DNA sequencing and developing a new statistical method, they found that selection has actually sped up.
Selection went especially bonkers during the Bronze Age (around 3,000 years ago).
That's when gene frequencies for everything from immune function to body fat to intelligence were most in flux.
Over the last 10,000 years, selection pushed the genetic predictor of cognitive performance up by roughly a full standard deviation — most of it between 4,000 and 2,000 years ago.
After we finished recording, David sketched out on a whiteboard his new heretical model about who the Neanderthals really were. Luckily, I took out my iPhone and managed to record it.
He thinks the standard story (that Neanderthals are some separate archaic lineage we interbred with a little) just doesn't fit the evidence. Instead, he proposes that Neanderthals are essentially genetically-swamped modern humans.
A small population somewhere around the Caucasus invented Middle Stone Age technology roughly 300,000 years ago and expanded outward. The ones that moved into Europe interbred with local archaic humans, got genetically swamped, and became Neanderthals. The same expansion went into Africa, met much more diverged archaic Africans, and that mixture became us.
This means Neanderthals and modern humans share the same cultural ancestry — the only difference is which archaic humans they mixed with afterward.
David is a brilliant and rigorous scholar. It was a real delight to learn from him again.
0:00:00 – Ancient DNA suggests strong selection over last 10,000 years
0:16:24 – Natural selection intensified during the Bronze Age
0:35:40 – Why didn't evolution max out intelligence?
0:58:00 – Evolution is limited by time, not population size
1:09:40 – Why no farming before the Ice Age?
1:17:52 – The Neanderthal puzzle David can’t stop thinking about
1:54:40 – The methodology behind this breakthrough
I'm pleased to share our latest preprint: Combinatorial effects of gene dosage, polygenic background and environment on complex traits 🧵https://t.co/KBpAJXm6nn
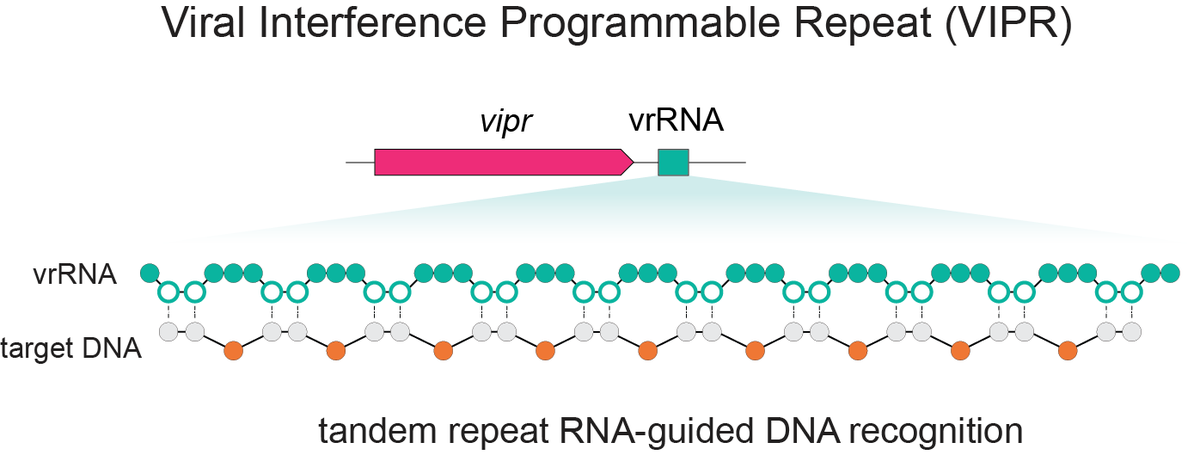
Excited to share our discovery of a new programmable RNA-guided DNA-targeting system hiding inside bacteriophages that predates CRISPR.
We call it VIPR (Viral Interference Programmable Repeat), and it uses an entirely new logic to find its targets.
Thread + link below.
Stoked for the day when the scientific standard for AI in Biology becomes “does it actually do the thing?” and not “does it point to a potential future in which it does the thing?”
Embryo selection using polygenic risk scoring has been a hot topic recently, with startups investing millions into the idea. Previous studies on embryo screening have reported relative risk reductions of up to 50%.
The high risk reduction estimates are, however, based on the assumption that each IVF cycle produces 2 to 5 viable embryos, all having a similar chance of a successful live birth. Sadly, that’s not the reality.
In a new preprint, the authors analyzed data from 6,944 real IVF cycles from 4,452 infertility patients. The reality of the IVF pipeline: on average, each cycle produced just 0.88 euploid embryos and 0.17 live births. You cannot select the lowest-risk embryo when most cycles don’t give you a choice.
Next, the authors simulated how disease risk reduction fares in this realistic scenario. The relative risk reduction ranged from under 0.5% across all cycles to just 1–3% in cycles that resulted in a live birth, far below the previously predicted 50%. It’s worth noting there is one setting where risk reduction reached a reasonable level of ~20%: egg donor cycles, where viable embryos tend to be many due to young donors. Even here, the estimate is a fraction of prior predictions.
The findings raise an important question: who is polygenic embryo screening actually for? It’s designed for patients with multiple viable embryos, all birth-ready, in a single cycle. Those patients don’t exist in most clinics.
The reality is that this technology is being built for healthy, fertile individuals with plenty of financial resources, doing IVF electively to have designer babies.
Klausner et al. medRxiv 2026
https://t.co/BQ5V344lFP
Really interesting new paper from Akbari et al. identifying a lot more selection in ancient DNA than previous approaches. I think it gets at three core challenges for this type of analysis where our understanding is still limited. 🧵
Nice article in the New York Times (https://t.co/i4KSK7zm2u) on Akbari et al. Nature finding a large number of novel loci under selection in ancient DNA data. Here's what I wrote when asked for comment:
A new paper in @Nature from David Reich, @aliakbari23 and colleagues breaks the conventional understanding of recent human evolution. The field believed that strong selection in the recent past (~10,000 years) was rare, with few exceptions like the lactase persistence locus. In this paper, the authors challenge that belief, showing that we weren't looking at the problem right.
Previous studies that looked for evidence of selection using ancient DNA addressed the problem cross-sectionally, asking if allele frequencies differed across populations more than what one would expect based on genetic drift and migration. Most arrived at the conclusion that population structure primarily explained the observed differences. Here, the authors addressed the problem longitudinally, accounting for when ancient individuals lived by explicitly modeling time as a variable in the analysis. It turns out doing it this way dramatically increases power, increasing the number of genome-wide significant selection signals by 20-fold!
Looking at why accounting for the time variable led to such dramatic changes in results, the authors find that previous studies missed so much because selection often happened not on new variants leading to dramatic sweeps (the conventional model: new variant -> selection -> increase in frequency) but on already existing variants driven by transient environmental pressures. Many of these variants underwent reversals, selected up when a pressure existed, then purged when it disappeared or the trade-off cost became dominant. A great example is the TYK2 variant, where an allele boosting immunity was selected for thousands of years because it protected against TB, then got purged as TB endemicity declined and the autoimmune cost took over.
The scale of what they found is striking: hundreds of loci showing strong selection in the past 10,000 years with a median selection coefficient of ~0.86%. This number is pretty big in evolutionary terms, meaning allele frequencies have been shifting by ~1% per generation in a consistent direction. Previous selection scans found a maximum of 20 loci, and this one finds hundreds. That isn't an incremental change. It fundamentally reframes our understanding of how common strong selection has been in recent human history.
Some of the most striking findings come from polygenic selection, where hundreds of small-effect alleles were pushed in the same direction simultaneously. Polygenic scores based on large-scale GWAS of today predict recent negative selection for traits like body fat, waist circumference and schizophrenia, and positive selection for others like cognitive traits. One important caveat is that GWAS phenotypes are measured in industrialized societies today, and how well they capture what was actually being selected in ancient environments is debatable.
For me personally, these findings have direct implications for drug discovery. When using human genetics to find drug targets, we often fixate on the benefit and risk profiles of variants visible today. But we need to be aware that a variant's benefit:harm ratio might be environmentally contingent, and could reverse when the wrong environment manifests. An evolutionary understanding of a variant's association with traits is therefore essential.
The same logic applies, perhaps even more urgently, to embryo selection. Selecting embryos based on polygenic traits is humans making permanent, heritable decisions for their offspring with a narrow view of today's environment. The ancient DNA record now shows that cost-benefit landscapes flip over time. So, an embryo carrying man-made selections is carrying those changes into an unpredictable future environment.
The broader takeaway is that human evolution didn't freeze in the last 10,000 years. We just lacked the tools and datasets to see its movement. The current findings are based on European populations. I am curious to see these analyses extended to other populations too, like South Asian, East Asian and African populations, which might be holding more surprises to blow our minds.
Akbari et al. Nature 2026
https://t.co/3WWjpTiVgA
I usually dump on EVO2 quite a bit (mostly since it's been overhyped to death beyond it's actual capabilities), but this is nice work showing that a better embedding approach with supervised probing can deliver strong performance on coding variant effect prediction. 1/
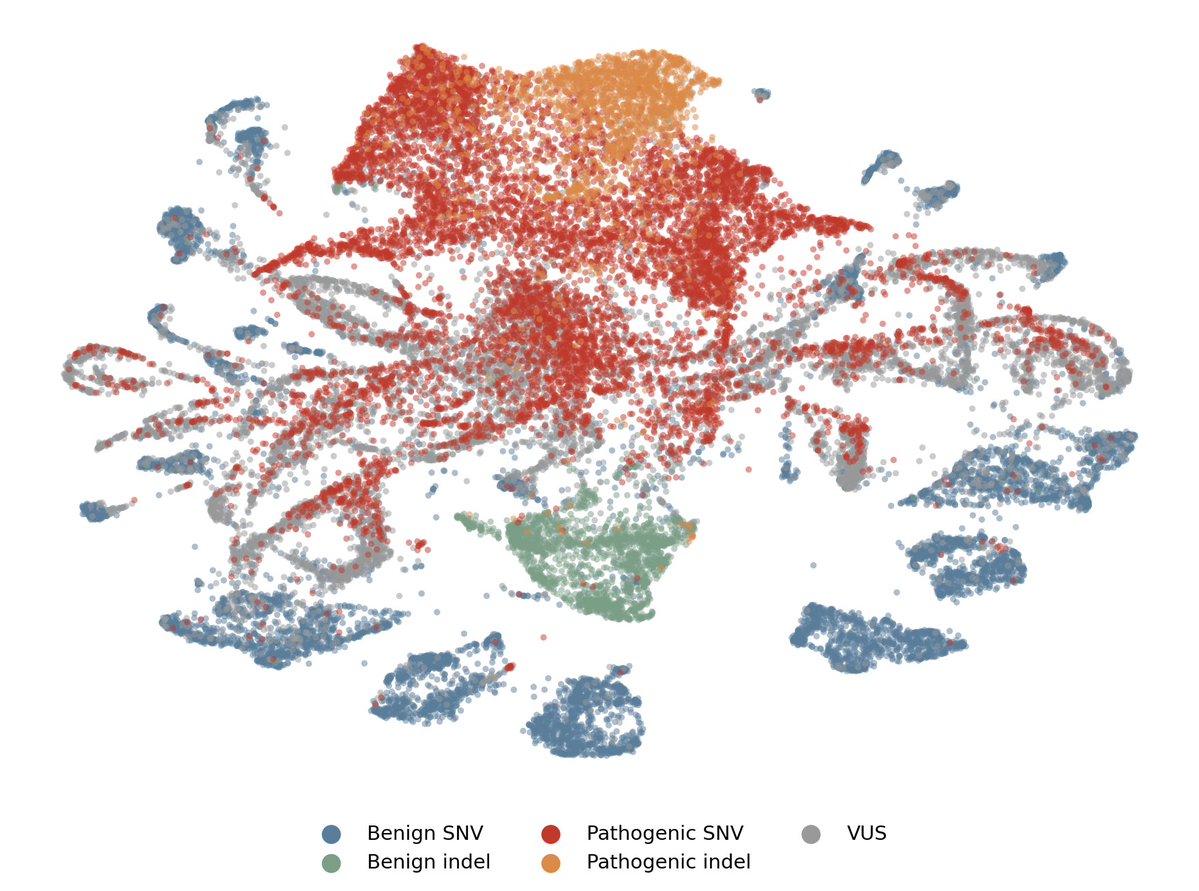
We achieved state-of-the-art performance in predicting which of 4.2 million genetic variants cause diseases by interpreting a genomics model, in a new preprint with @MayoClinic.
We're now releasing an open source database for all variants in the NIH's clinvar database. 🧵(1/8)
Happy to see our work on repeat expansions led by my colleagues--Sahar Gelfman & Vijay Kumar--at RGC published in @Nature.
Below is a thread highlighting what we learned from systematically studying repeat mutations at population-scale in ~1 million individuals
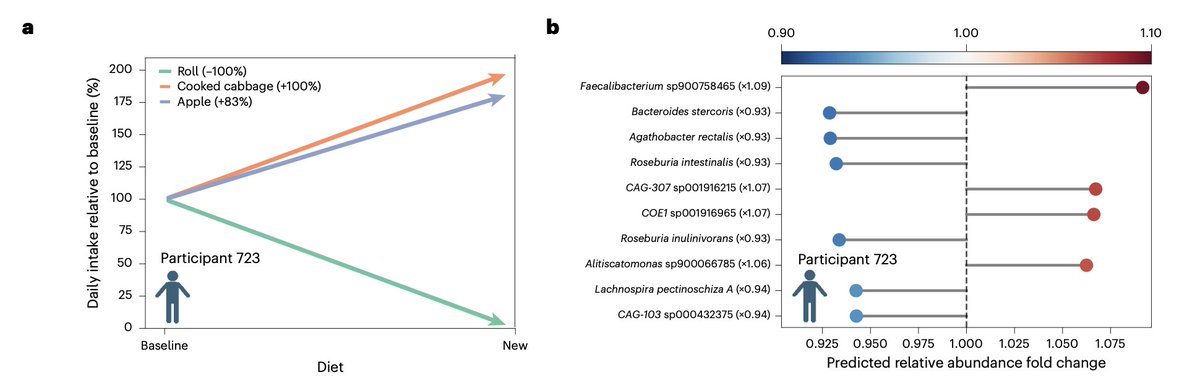
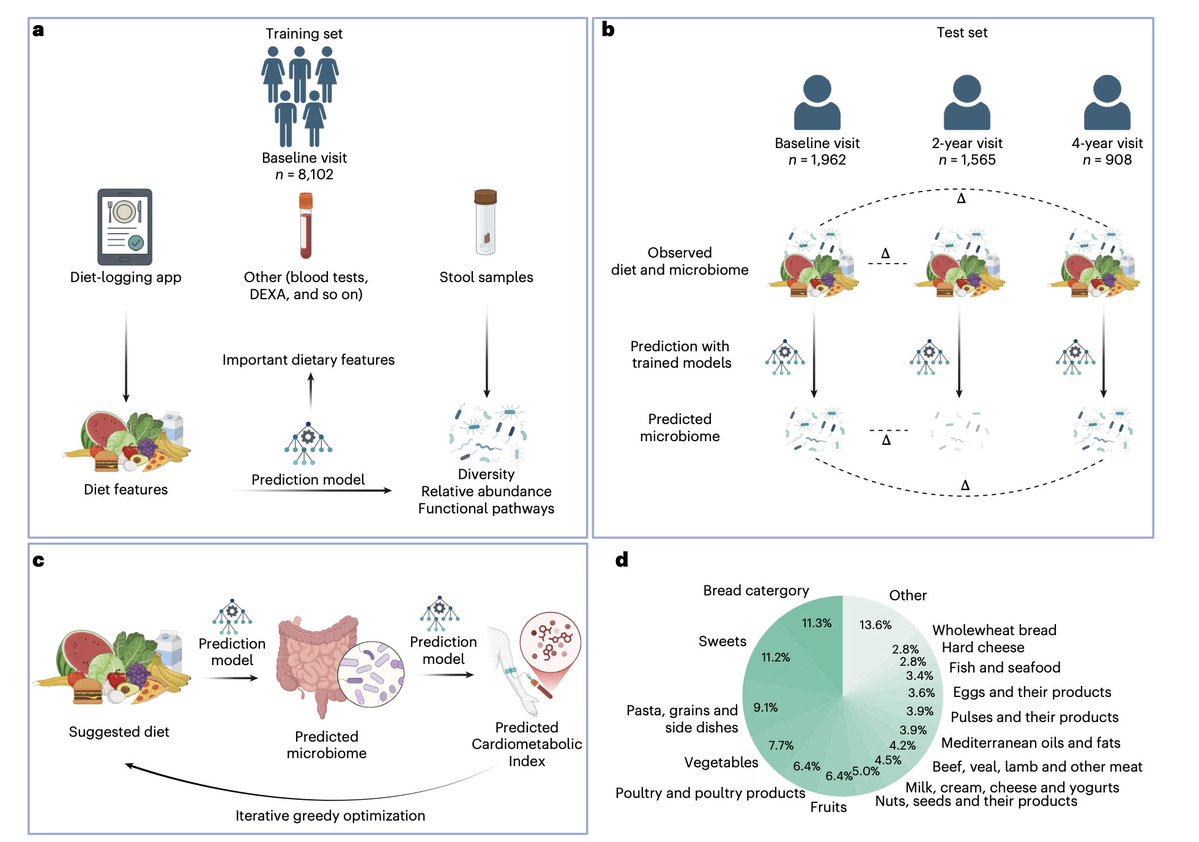
Our new study @NatureMedicine from the Human Phenotype Project: Analyzing diet and microbiome data from 10,000+ people, we found that what you eat is strongly linked to which microbes live in your gut, down to specific foods like coffee, yogurt, and milk driving distinct microbial signatures. We also simulate personalized dietary interventions with predicted microbiome shift effects that are associated with improvements in cardiometabolic health
Read here: https://t.co/JoIEoIqJ6O
HPP: https://t.co/sdxsQpoStl
There is a reason I have consciously stayed clear of training and using large AI models in biology before understanding precisely how & what smaller incarnations learn. Here is the fundamental problem 1/
New AI paper from us this week. When my student first showed me his initial findings, I really didn’t know what to make of them. I felt that this was an interesting but curious loophole phenomenon that would shortly be closed. I was very wrong.
https://t.co/H3YIyl01FR
Exome sequencing and analysis of 44k South Asians from the Genes & Health cohort (G&H) now published in Nature Genetics.
Of all the South Asian centric cohorts, G&H is my favorite. They have been doing high quality work and sharing the results generously to everyone in the field.
G&H is dedicated exclusively for British South Asians of Pakistani and Bangaldeshi heritage. In the current work, the authors studied whole exome sequencing (WES) of 44,028 individuals.
The value of such cohorts become immediately evidenct when you contrast with existing large European-based cohorts/databases such as UK Biobank, gnomAD. Some interesting stats reported in this paper:
- 26% of G&H coding variants are completely absent in gnomAD (this rises to ~50% when compared only with the European subset of gnomAD)
- 33% offspring of consanguineous marriages (vs 2% in the UKB)
- consequently, 1.8% of average genome is in runs of homozygosity (ROH) (vs 0.4% in UKB)
- this high autozygosity yields 2.4x more homozygous genotypes than what you'd expect under random mating
- for ~3k genes at least one homozygous (or comp het) pLOF carrier ('knockouts') was found, of which 1,669 are seen in only G&H despite being 10x smaller than UK Biobank.
For 546 genes, knockouts are seen only in G&H when compared to even 5 major larger biobanks combined (~1.4 million)
Great work and an incredible progress in South Asian genetics, again, unsurprisingly from a South Asian cohort built outside South Asia
Congrats to G&H team!
Kim et al. Nat Gen 2026
https://t.co/bwrwFZeOKd
The world would be a much better place if every LLM contribution was watermarked and I think we as a society will greatly regret rushing into unsigned AI.
Today we're launching Latent-Y: the world's first autonomous agent for drug design, lab-validated end to end.
Give it a research goal. Latent-Y reasons, designs, iterates, and delivers lab-ready antibodies, autonomously or collaboratively, with the biological reasoning of a PhD protein design expert.
Technical report: https://t.co/E7IHfkvvD3
Blog post: https://t.co/GfJAfzj0Qx
Apply for access: https://t.co/E0SR9znZiP
Great to the see the flurry of single gene knockdown Perturb-seq like atlases from cell-lines, mouse brain etc over the last few days. These are undoubtedly very valuable datasets. I just want to re-iterate a few other very important expt. design considerations 1/