@anniepaul@jennfrey Scientists already deal with a flood of easy-to-ignore low-quality papers, so more slop on PubMed and arxiv is unlikely to be a problem. The bigger danger is AI-massaged results that risk tarnishing the reputation of unwitting collaborators and PIs and institutions
1\ I ran OsIPI1, a rice RING-type E3 ubiquitin ligase that's ~88% intrinsically disordered, through 4 state-of-the-art structure predictors. Same protein, same MSA. The results are wildly different. Here's what I found:
“A university that asks students what they want and gives it to them is not educating – it is catering.”
In my annual MBZUAI commencement address today, I emphasized two principles that shape our university: 1) The core business of university is creating knowledge and teaching knowledge, everything else is secondary; 2) Our university is a forge. Faculty and leaders are not pastoral counselors; they are forge masters. Their job is to make people capable, not comfortable. — Maybe obvious, but hard to say and hard to enact these days.
https://t.co/nq6YjFUP0T
@TimothyDuignan@SynBio1 Solving the true folding problem requires us to have clear benchmarks on what it means to have solved it. These don't exist (yet).
Based on lots of developer feedback, we just updated the PPIFlow codebase. We've unified the installation environment and simplified the entire pipeline to make it way easier to use. Hope this helps everyone get started faster!
Check it out https://t.co/yLmmzaHNoC
Based on lots of developer feedback, we just updated the PPIFlow codebase. We've unified the installation environment and simplified the entire pipeline to make it way easier to use. Hope this helps everyone get started faster!
Check it out https://t.co/yLmmzaHNoC
@MartinPacesa@DdelAlamo and are largely concentration-independent. On avidity: we immobilized His-tagged VHHs and measured binding to monomeric targets. Loading stayed <1 nm, and Rmax values (0.2–1 nm) were consistent across concentrations.
It would be very helpful if better BLI setups can be provided.
@MartinPacesa@DdelAlamo Thanks for commenting on this point. Our analyte concentrations are in line with recent work (e.g., RFdiffusion etc.). We kept ligand loading moderate (~0.6 nm) to limit surface crowding. For high-affinity binders, dissociation curves show clean exponential decay (no plateau)
Something about the PPI-Flow paper that raises an eyebrow is its claim of 100% expression success in both minibinders (cell-free synthesis) and VHHs (CHO cells), despite relying on ProteinMPNN/AbMPNN for sequence design, which has shown issues in other studies
The proteina-complexa paper is the 2nd this month suggesting that high K+E content, a staple of ProteinMPNN designs, is predictive of poor expression (the bits in bio benchmark paper showed a few weeks ago). In their case, they measure poor sequence recovery in phage display
I usually don't like to criticize papers on social media, but this one deserves it. Not familiar with @Ligandal, but so many problems: AI-hallucinated citations, figures, no real validation, not "structure-free", and definitely not diffusion. I'll go thru my criticisms below. 👇
Really great collaboration between @ManifoldBio and @nvidia to test 1M de novo designed binders against 127 targets, measuring over 100 million potential protein-protein interactions! This was a great collaboration with some very exciting results. NVIDIA's new Proteina-Complexa method is SOTA for de novo minibinder design. If you're interested in designing minibinders to targets you couldn't hit with other methods, try it out!
I'm particularly excited about what this large scale data enables. As we generate 1000s of experimentally validated structures, this data becomes the input to training new protein design models. At Manifold, we are generating datasets of this size continuously, and have experimentally validated thousands of de novo designed binders across many formats (VHH, minibinders, peptides, etc). New models will open up new hard to hit targets, paired with our large scale in vivo measurement, will enable us to create previously impossible therapeutics.
Up next is training new models on this and other data we have generated, stay tuned! And thanks to NVIDIA for setting up such a great collaboration, its been fun and fruitful!
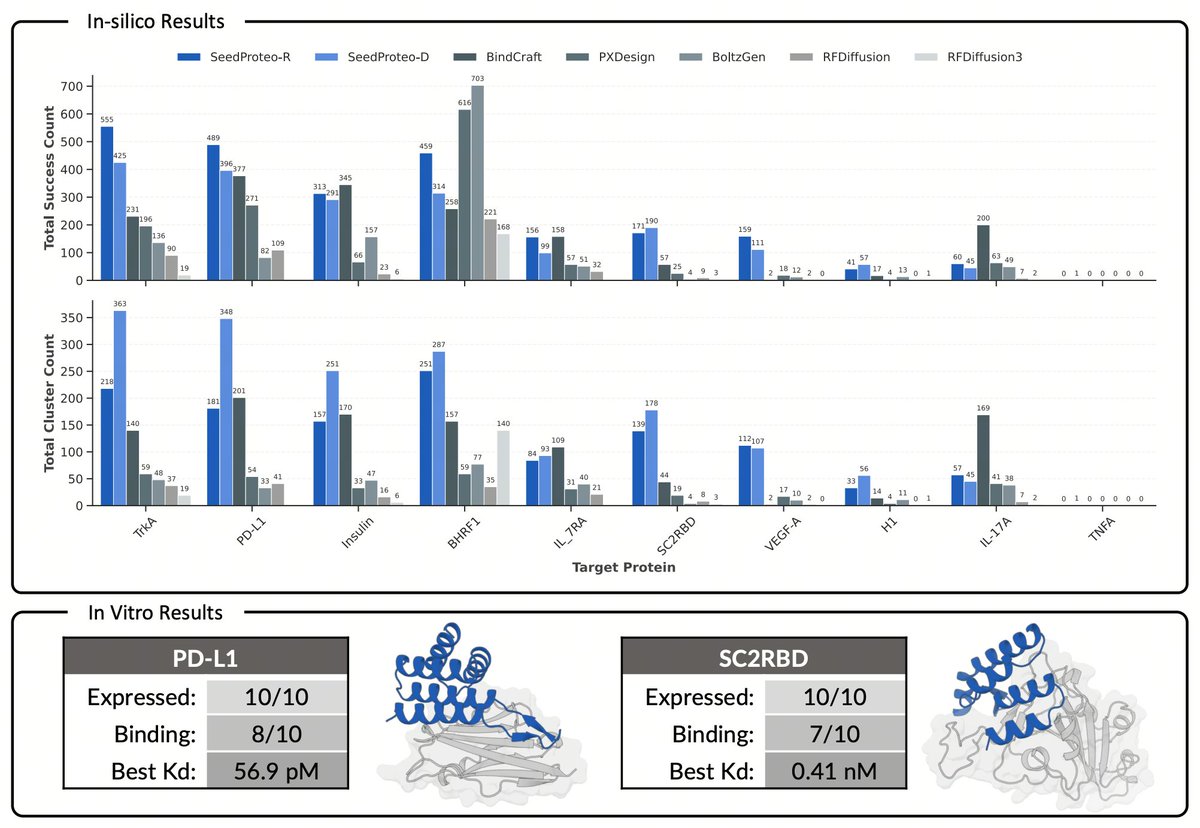
Here is SeedProteo, our latest diffusion-based model for de novo all-atom protein design from ByteDance Seed! Our server is now live — feel free to give it a try! https://t.co/Tqsu2VPmWw
@AsimovPress@btnaughton You forgot to mention PPIFlow: it's open-source, includes detailed documentation and carefully benchmarked results, along with experimental validations.
Link: https://t.co/XHpYJRA2x4
@AsimovPress@btnaughton You forgot to mention PPIFlow: it's open-source, includes detailed documentation and carefully benchmarked results, along with experimental validations.
Link: https://t.co/XHpYJRA2x4