What can you describe in a VCF? A haplotype, an allele, a diplotype, a set of annotated sites, ... HGVS is the same way.
It would be much better if these instead described *one thing* (i.e. atomic variants) and another described the composition of those things (i.e. diplotypes)
@iskander EBV is pretty standard (it's in all our human references as a decoy) because immortalized EBV is extremely common in human samples. I'm less certain about the others. (We do metagenomic screening on every sample and I don't recall seeing these, but I could go check...)
This is a great question! Getting consistent results out of RNA assays is stupendously difficult. Here's what we currently do, and what we plan on doing in the future:
1. We have our own full-length transcript capture assay that dramatically improves the number of reads that are informative with respect to splicing analysis. Short reads generally capture zero or one splice junction each. A single splice junction does not uniquely identify a transcript; only full-length reads can do that reliably. (Right now about half of the reads we put through a sequencer have both the 5' TSO and 3' poly-A tail we're looking for. We are actively investigating ways to increase that percentage.)
2. Because of (1), we do not do imputation of transcript counts. Instead, we output minimum and maximum values for transcripts based on the observed transcripts (which may sometimes be incomplete). You can impute counts from these if you'd like.
3. We do interleaved replicates to help control for batch effects. (Even things like the ambient temperature in the lab has observable effects, so you really need to run your controls in tandem.) Moreover, we run the same small number of *automated* protocols over and over in our lab, so it's easier for us to control for more environmental factors.
4. Our long-read RNA aligner is designed specifically for aligning (short or long) spliced reads. It can correctly align to small exons (as short as 12 bases, like STAR) but works fine with very long reads (hundreds of kilobases) and produces base-level affine-gap alignments (like minimap2). The output of our aligner includes information about the splicing status and 5' cap / 3' poly-A tail that isn't normally conserved, and you can see those in our web viewer.
Chemistry improvements that we're currently researching:
1. Selectively isolating m7G-capped transcripts so we can distinguish between nascent and mature RNA and reduce the number of reads that are caused by intronic priming of pre-mRNA on poly-dT probes.
2. Move away from template switching, which has bias, poor efficiency, and occasionally truncates transcripts.
3. Synthesizing our own RNA ladders in order to test the repeatability and efficiency of our isolation techniques for different kinds of transcripts. (Currently we just test against bulk RNA standards, which do not tell you much about biases in your protocol.)
@basedsystems "Alternative splicing analysis with no bioinformatician needed"
Given that any two splicing tools will be wildly discordant, why should we trust your platform?
The basic architecture we use for aligning long RNA reads is a seed chaining algorithm (similar to minimap2) on top of a suffix array (like STAR). Suffix arrays have slightly better sensitivity for short exons than minimizers, so it was worth the perf trade-off over minimizers. Obviously the seed chaining scoring has to be modified slightly because now seeds are variable-length exact matches from the suffix array, and of course you have to handle "deletions" near splicing signals with special care.
Our suffix array contents are also gene-locus-aware, so it preferentially produces sense-oriented matches. (We only try anti-sense-oriented matches when less than 80% of the read bases can be lined up to a sense-oriented gene locus.) This ends up being important for transcript assignment when you have partially-overlapping loci that have different orientations, which is quite common.
We can also shove barcodes like CITE-seq probe sequences into the suffix array and use those as dummy "loci" for transcript counting purposes. The downstream stooling just treats these as additional gene counts that can be filtered / projected as usual.
One final bit of special sauce is that for full-length RNA chemistries that have indexes attached to the 5' cap and 3' poly-A, we're able to use that information to narrow down which specific transcript in the target locus best matches the read.
We wrote our own hyper-efficient version of the STAR aligner.
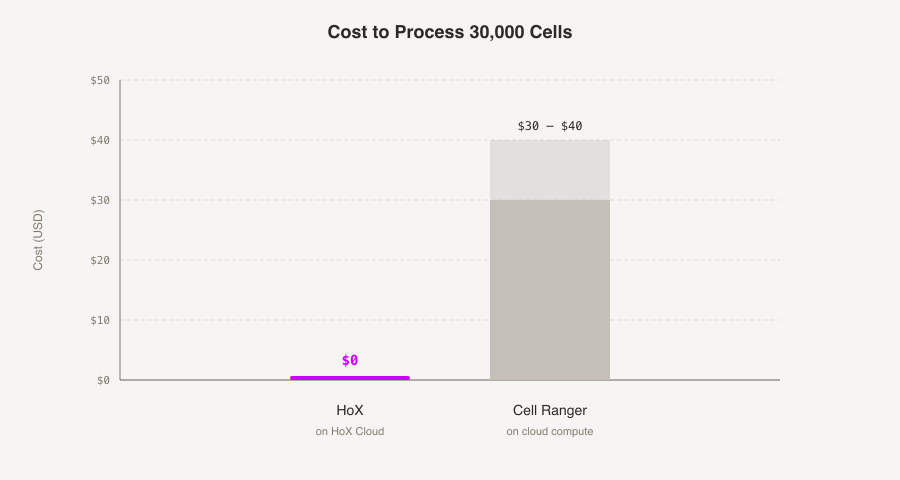
Typically it costs around $35 in cloud compute to process 30k cells with Cell Ranger. Ours costs so little that we don't charge for it. Also ours works with nanopore.
If you prefer pseudoalignments, then you can run it in pseudoalignment mode -- obviously we don't charge for that too.
All results instantly integrated with our other tools. As soon as a run finishes you can open alignments or expression matrices in our viewers without moving anything.
Let me know if you want to try it!
@NmrReyez@m_goes_distance@shelbynewsad That's an assay for MRD targeting SNVs; the Ultima reads generally are quite specific for SNVs so no issues there. Doesn't solve the performance issues on indels, though.
@NmrReyez@m_goes_distance@shelbynewsad It works great for most short-read applications *except* germline genotyping. Even with Ultima's own re-trained DeepVariant, performance on homopolymer indels is very poor.
Most people I know in AI think the median person is screwed, and they have no idea what to do about it.
I spent the last 3 months talking to dozens of researchers, economists, and policy experts about AI's impact on work; including reps from every frontier lab and several Congressional offices. Unfortunately, I was not reassured.
The AI industry is raising the alarm, but can't change course. These companies' core business model relies on the disruption they are warning about: their faith in full automation only makes them go faster.
Policymakers are waking up, but still paralyzed by data and debates. Econ wonks disagree on plenty, but even the limited scenario looks like a "painful transition" that will disempower millions of workers.
But an "underclass" is not inevitable, but rather a societal choice — and one we can and should stop. Instead of waiting for impact, we should start planning now to support workers through AI disruption. Whether policymakers can assuage concerns about economic security may determine if we get to reap AI's gains at all.
New from me for @NYTOpinion. I put a ton into researching what I think may be the biggest topic of the year, so hope you read it (gift link here!) https://t.co/NiGJpjyjzH
@shelbynewsad At least according to Ultima, yes they've had $100/genome for a while. Caveats are the usual ones: you need to have volume pricing on reagents and 100% utilization of the wafers.
(The other Ultima-specific caveat was/is that homopolymer indels are very common in their reads.)
Great question - these are commonly used in the biotech industry. So to answer your original question of whether there’s a way to make money doing this, the answer is no
Concretely: the A100 in the P2I has a perf/$ for base calling that is roughly 20x worse than a 5080. So we run a bunch of 5080s. (The pacbio revio has A100s in it too, by the way...)
I don't want to be forced to purchase a "sequencer" that bundles a bunch of obsolete GPUs. I just need the sequencer part! I have racks and racks full of compute already.
Everyone I've talked to who operates a P2 Solo is bummed out that ONT is discontinuing them. They are, by quite a wide margin, the best machine that ONT makes (IMHO).
Robot arms are the lab automation equivalent of a "code smell." The ideal number of humanoid robot arms involved in your protocol is zero (because there's nothing about human biomechanics that makes us good at running microbiology assays...)
We use one arm to load and unload liquid handlers and to move plates to our plate reader, but we're pretty aggressively trying to move stuff entirely "on-deck." I'll be perfectly happy once we can throw out the arm entirely.
@iskander@josiezayner We have a solution for RNA selection and stabilization figured out so you don't need to ship on dry ice (do first strand synthesis in the collection kit tube!) but even if you ship same-day to a sequencing site you're probably looking at 48hr from collection to answer.
@iskander@josiezayner We've thought a lot about the clinical metagenomics angle since we already have a pipeline for environmental samples, but getting ultra-fast turn-around in the end-consumer setting is tough.