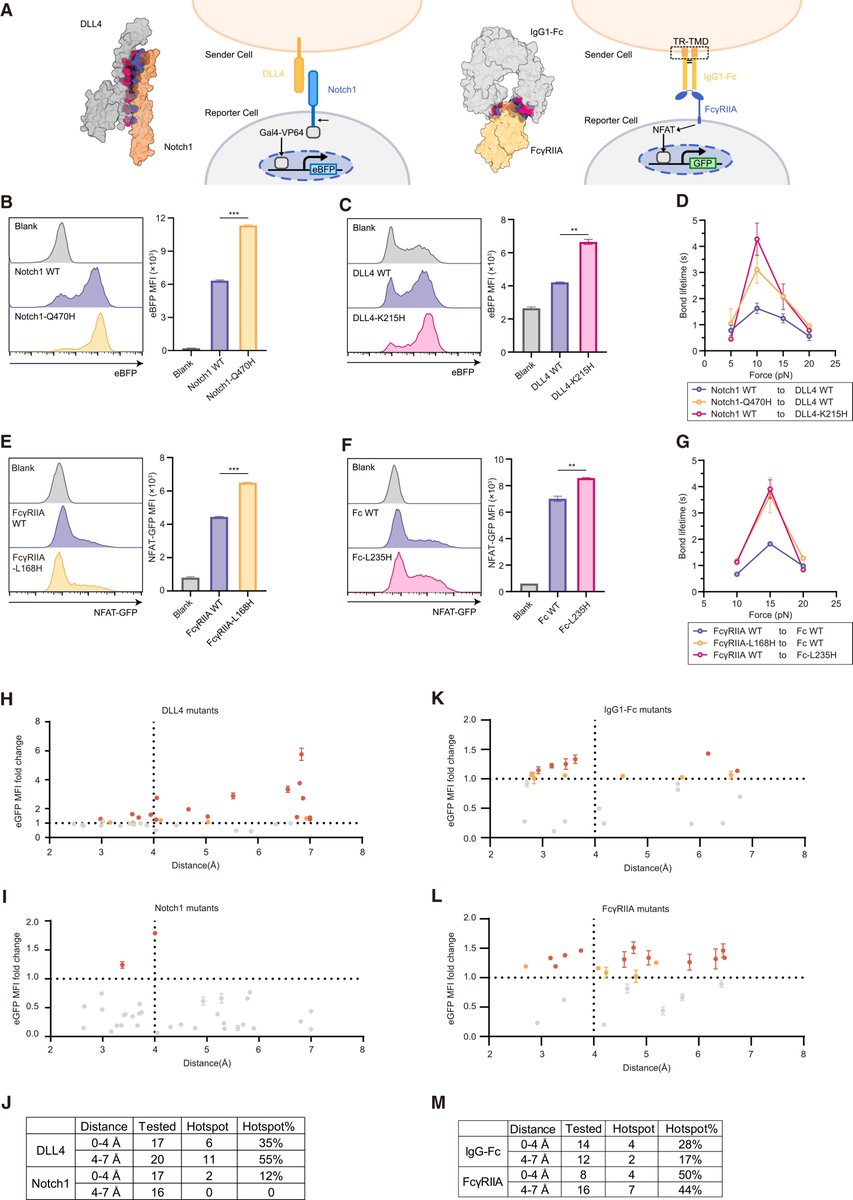
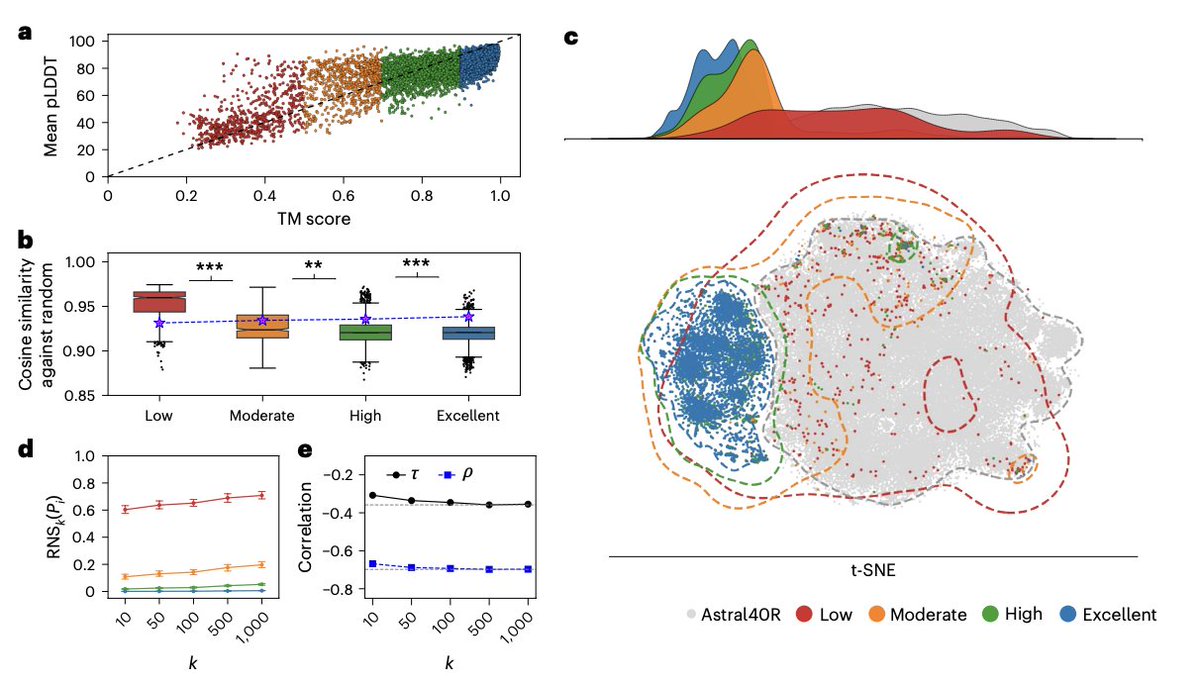
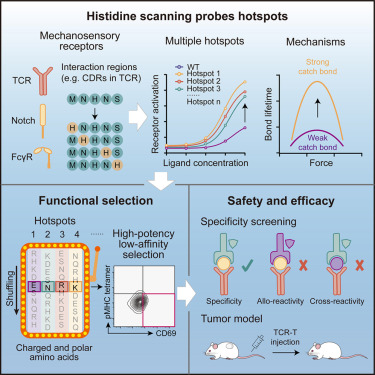
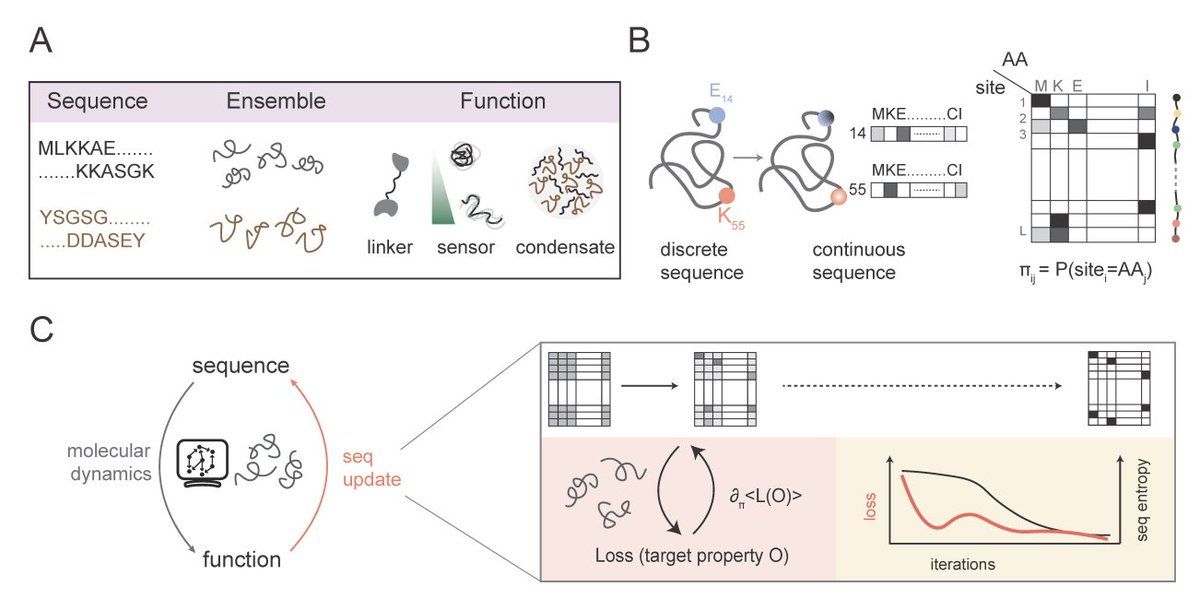
Very happy to see our work published!
Synthetic data power!
Thanks for @NatComputSci for their support and to the 5 positive reviewers for feedback!
Thanks to @chevaliersf for writing a very cool News & Views about it too!
Now, Enjoy!
. @pandaisikit, @probertimmodels, @victorgreiff and colleagues introduce the Absolut! framework, which can generate synthetic 3D-antibody-antigen structures to assist machine learning and dataset construction for antibody design. https://t.co/OaSbqtF5Pb
👉https://t.co/7GTf7zcvUA
Very interesting... We rely on embeddings generated from similarity loss, then well, these embeddings mess up similar sequences that are functionally different. Makes sense ;p
When a protein embedding is indistinguishable from noise
Protein language models have become the backbone of computational biology. Feed them an amino acid sequence, and they return a dense vector—a compact numerical fingerprint that downstream models use to predict function, structure, localization, or the effect of a mutation. The assumption, largely unquestioned, is that this fingerprint actually encodes meaningful biology.
Prabakaran and Bromberg challenge that assumption directly. They ask a deceptively simple question: how do you know whether a given embedding actually represents a protein—or whether it's just noise dressed up as a vector?
Their answer is the Random Neighbor Score (RNS). The idea is elegant: generate biologically meaningless sequences by randomly shuffling the residues of real proteins—preserving amino acid composition but destroying all evolutionarily meaningful interactions. Then, for each real protein, measure how many of its nearest neighbors in latent space are these random imposters. A high RNS means the model never learned to place that protein somewhere biologically meaningful.
Applied to ESM-2 and ProtT5 across thousands of proteins, RNS correlates strongly with structural prediction quality: proteins with poorly predicted structures have embeddings nearly indistinguishable from random sequences. Downstream tasks follow the same pattern—contact prediction precision drops roughly 40% for high-RNS proteins, and variant effect prediction falls to near chance. Most sobering: between 19% and 46% of the human proteome is underlearned by current models, depending on architecture. Intrinsically disordered regions fare especially poorly across all architectures tested.
RNS is model-agnostic and computationally cheap—around two minutes on GPU for 10,000 proteins—making it a practical prescreening step before any embedding-based inference.
For R&D teams that routinely use protein embeddings to prioritize variants, annotate novel sequences, or screen large libraries, this has immediate consequences. Running RNS before downstream inference flags proteins where predictions are unreliable, reducing the risk of propagating errors into expensive wet-lab campaigns. It also offers a principled way to identify gaps in model coverage—directly actionable for teams building or fine-tuning their own foundation models.
Paper: R. Prabakaran & Yana Bromberg, Nature Methods (2026) — CC BY-NC-ND 4.0 | https://t.co/JOblk7kT6R
Is it reasonnable that big tech invest 1000 euros per human on the planet on big data centers... Sounds to me that the AI become more expensive than humans, and that half of this money could have made more people happy than the AI users (unless this money is a bubble)
Evaluating AI-generated drug molecules: when 3D shape matters as much as chemistry
AI models can now design drug-like molecules from scratch, placing atoms in 3D space to fit snugly inside a protein's binding pocket. But there's a catch: many generated molecules adopt physically impossible shapes—atoms crammed too close together, rings twisted unnaturally, or bonds frozen in high-energy orientations. These aren't minor cosmetic issues; they mean the molecule couldn't actually exist as designed.
How do you check millions of AI-generated structures for physical plausibility? Geometric rules (measuring distances and angles) are fast but miss anomalies nobody thought to define. Quantum-mechanical calculations are accurate but far too slow. We need something in between.
Fan and coauthors propose an elegant solution: use AI to evaluate AI, through two complementary tools.
The first, HEAD (High-Energy Atom Detector), computes the energy of every individual atom using a machine learning force field. The logic is powerful: any geometric anomaly—clashes, distortions, misplaced atoms—shows up as abnormally high atomic energy, even if that specific error type was never explicitly defined. Comparing each atom's energy against thresholds from known valid molecules, HEAD catches problems that geometric checks miss, 30× faster.
The second, TED (Torsional Energy Descriptor), examines each rotatable bond after basic structural cleanup. Even a geometrically sound molecule can have bonds stuck in energetically costly orientations—something drug designers care about deeply. TED predicts full rotational energy profiles using a neural network trained on millions of molecular fragments, achieving near-quantum-mechanical accuracy at a fraction of the cost.
Testing five recent generative models across 102 protein targets reveals that no model excels at everything, and only ~20% of molecules from the best models survive all quality filters. Strengths are complementary: some models build better shapes, others handle bond rotations more realistically.
The takeaway extends beyond drug design: as generative AI enters more scientific domains, we need evaluation tools as sophisticated as the generators—fast, physically grounded, and precise enough to tell developers exactly what to fix.
Paper: https://t.co/YkHf1pxXvC
To celebrate the 1st November, I hope Sarkozy will also stay 1 more day per year in jail like the "solidarity day" where he forced people to work 1 day more for free every year!
When protein design meets differentiable programming
Most protein design tools assume a stable 3D fold. But many biologically critical proteins are intrinsically disordered: they never adopt a single structure, instead flickering across vast ensembles. Standard ML approaches that predict a folded structure don’t apply.
Ryan K. Krueger, Michael P. Brenner, and Krishna Shrinivas present a differentiable framework that inverts molecular simulations. The key idea: represent a sequence as a continuous probability distribution over amino acids, run coarse-grained simulations to model its ensemble properties, and then backpropagate gradients directly through the physics. This turns sequence design into an end-to-end optimization problem, where objectives can be tuned for size, flexibility, responsiveness, or binding.
With this method, the authors design disordered proteins that act as loops or linkers, remain compact yet disordered, or function as sensors that expand or contract in response to salt, temperature, or phosphorylation. They even extend it to create candidate binders for other disordered targets—long viewed as one of the hardest problems in protein engineering.
The result is a general recipe for physics-grounded differentiable design: keep the molecular simulator, make it differentiable, define the right loss, and let optimization explore sequence space efficiently. For applied ML, it’s a blueprint showing how simulation and differentiable programming can merge to tackle problems beyond text or images—pushing generative design into the messy, high-dimensional space of biology.
Paper: https://t.co/us3iRqUKig
correlation, association, hazard or causality?
Gipsies ;
copper stolen ;
no signal on railway ;
train blocked in a cow field ;
big delay :p
and it's even not Deutsche Bahn!
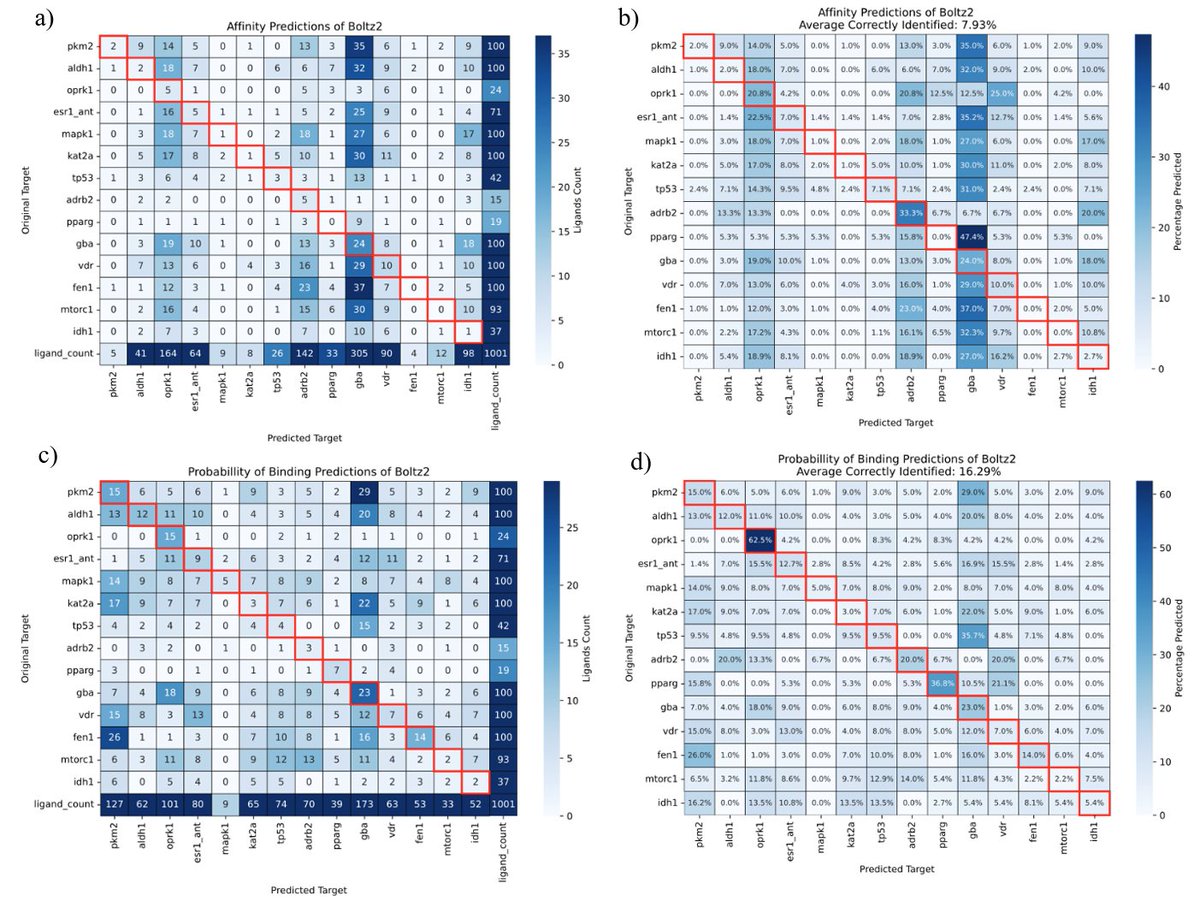
DL affinity model inter-protein scoring noise problem:
When comparing binding affinity predictions across proteins, scores aren’t calibrated on the same scale.
Example: On IVS (1,001 actives / 14 proteins), Boltz-2’s top-1 target ID ≈ random, with strong pocket bias.
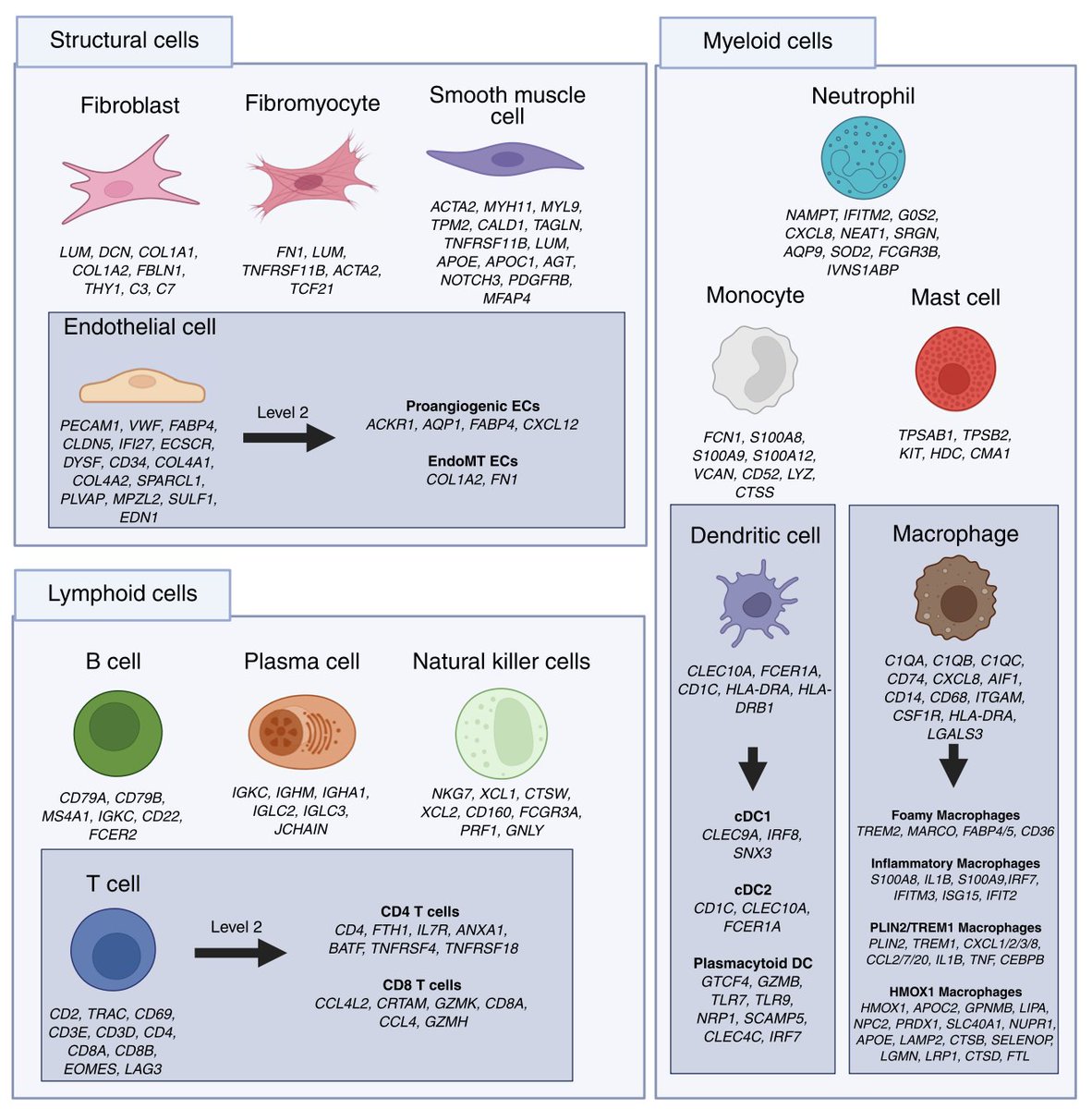
A very comprehensive effort to develop a single-cell atlas of human atherosclerosis based on 79 plaque samples from 3 vascular beds and >250K cells👇
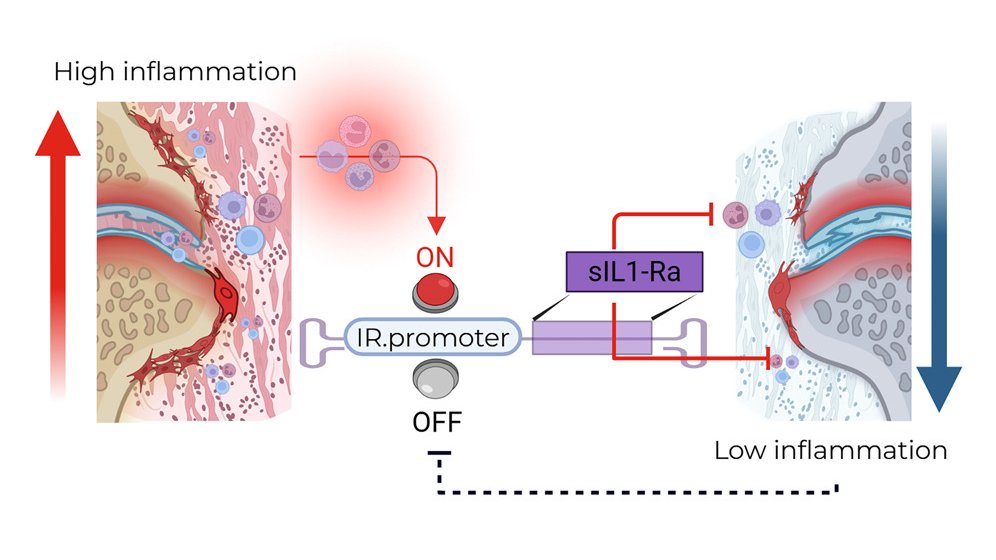
Inflammation-inducible IL-1-targeted therapy using an rAAV vector as a long-lasting, pathophysiologic treatment for chronic inflammatory diseases @MolTherapy@UMassChan 🇺🇸
https://t.co/I0Cy2BfLyC
A new benchmark for deep learning based affinity prediction: Solving the inter-protein scoring noise problem
1. Researchers from the University of Münster have introduced a novel benchmark for evaluating deep learning models' ability to predict protein-ligand binding affinities, addressing the inter-protein scoring noise issue that plagues classical scoring functions. This benchmark aims to assess whether models can accurately identify the correct protein target for a given active molecule, a critical capability for reliable binding affinity prediction.
2. The study utilized the LIT-PCBA dataset, which contains known active compounds for various protein targets, to create an inverse virtual screening benchmark. By applying the Boltz-2 model to predict binding affinities across all targets, the researchers tested its ability to distinguish the correct target among decoys. Surprisingly, despite Boltz-2's claims of high accuracy, it failed to consistently identify the correct protein target, highlighting the ongoing challenge of generalizing protein-ligand interactions.
3. The results showed that Boltz-2's predictions were not significantly better than random selection, with only a few targets showing enrichment of correct predictions. This suggests that the model may struggle with the variability in binding pockets across different proteins, a problem analogous to the inter-protein scoring noise in traditional scoring functions. The findings emphasize the need for more robust and generalizable models in the field of protein-ligand docking.
4. The study also explored various settings and parameters of the Boltz-2 model, including different diffusion sampling steps and molecular weight corrections, but none of these adjustments led to substantial improvements in the benchmark performance. This indicates that the issue may be rooted in the model's fundamental approach to predicting binding affinities, rather than specific computational settings.
5. The authors concluded that while deep learning models like Boltz-2 have shown promise in certain applications, there is still much work to be done to achieve accurate and generalizable binding affinity predictions. The proposed benchmark provides a valuable tool for evaluating and improving future models, emphasizing the importance of addressing protein flexibility, desolvation, and other factors that influence protein-ligand binding.
📜Paper: https://t.co/Wo0EZqpV8Y
#DeepLearning #ProteinLigandDocking #Benchmarking #BindingAffinityPrediction
Julia language could have been great...
If they didn't let people make packages with 2000 incompatible versions like in python.
Even ChatGPT gets confused in syntaxes that were only valid once when pluto aligned with mars.
TorchANI-Amber: Bridging neural network potentials and classical biomolecular simulations
1. The introduction of TorchANI-Amber marks a significant stride in integrating machine learning potentials with traditional molecular dynamics simulations. This interface seamlessly incorporates ANI-style neural network potentials into the widely-used Amber software suite, supporting both sander and pmemd engines. It not only implements all published ANI models but is also extensible to other energy predicting potentials through a simple mechanism, requiring no knowledge of Amber’s codebase.
2. One of the core innovations of TorchANI-Amber is its optimized CUDA implementation for computing the ANI models’ feature vectors. This enables simulations of systems with hundreds of thousands of atoms at the neural network’s level of theory, approaching DFT accuracy. The interface is designed to be fully compatible with all Amber capabilities, allowing users to leverage ANI potentials instead of traditional force fields for large biomolecular systems.
3. The study demonstrates the stability and energy conservation of ANI potentials through molecular dynamics simulations on various biomolecular systems, including ubiquitin and Trp-cage proteins in explicit solvent. The results show that ANI models can maintain molecular connectivity and structural stability throughout the simulations, with comparable energy conservation to classical force fields.
4. TorchANI-Amber also showcases its versatility in enhanced sampling techniques. The T-REMD simulations of Met-enkephalin in vacuo illustrate the compatibility of ANI potentials with well-established enhanced sampling methods. The increased diversity of sampled conformational space under T-REMD dynamics highlights the potential of ANI potentials for exploring complex biomolecular systems.
5. Performance benchmarks reveal that TorchANI-Amber achieves high performance on large systems, with ANI ensembles capable of running at up to 1 ns/day for systems of 25,000 atoms. The interface also supports the Amber-NB scheme, which offloads inter-molecular terms to the underlying MD engine, maintaining comparable performance while addressing the short-range nature of NN-IPs.
6. Future work on TorchANI-Amber will focus on refining the ANI architecture to include long-range interactions and extending the interface to support multi-GPU evaluation. This will further enhance the capabilities of ANI-style potentials within Amber and expand their utility for simulating large biomolecular systems.
📜Paper: https://t.co/URbI0ucSVX
#TorchANI #Amber #NeuralNetworkPotentials #MolecularDynamics #BiomolecularSimulations #MachineLearning
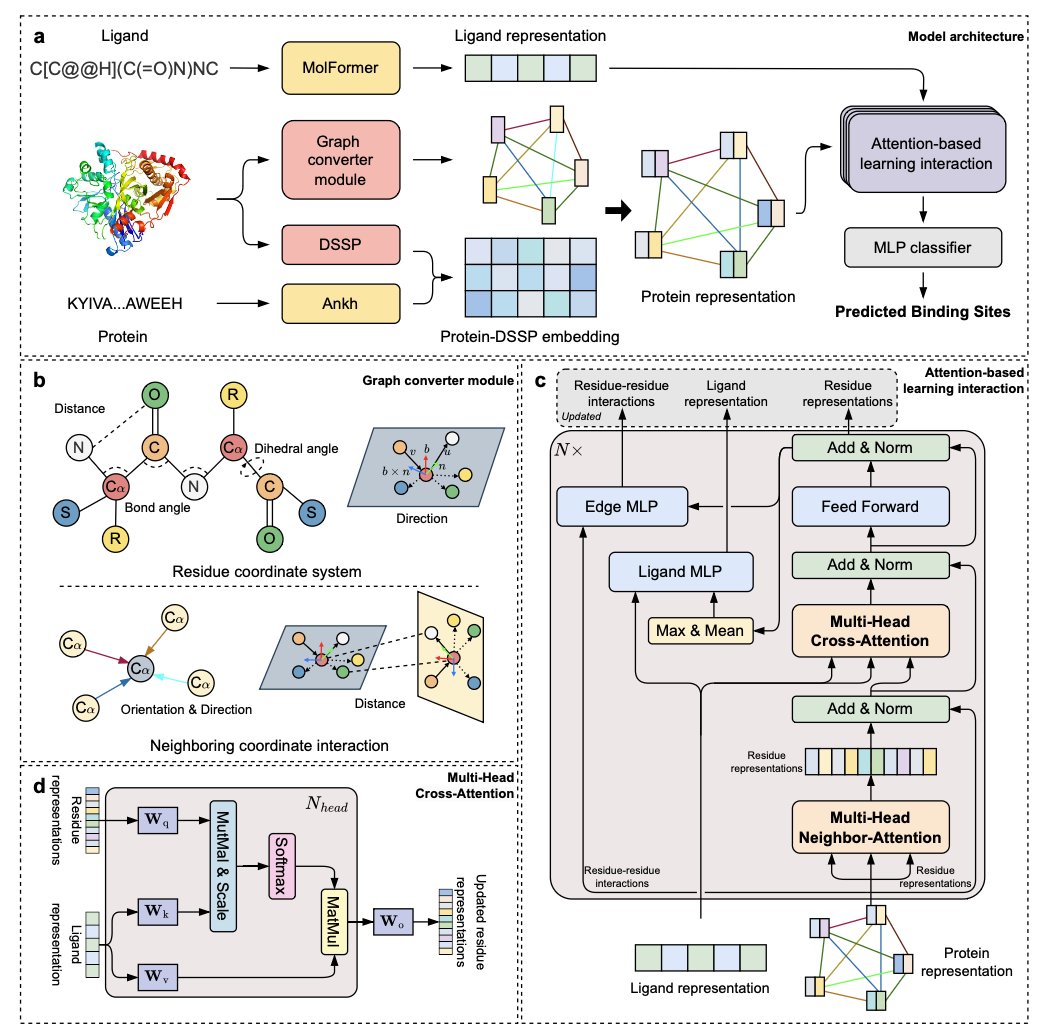
LABind: Identifying Protein Binding Ligand-Aware Sites via Learning Interactions Between Ligand and Protein @NatureComms
1. LABind is a novel structure-based method that predicts protein-ligand binding sites in a ligand-aware manner, utilizing a graph transformer and cross-attention mechanism to capture binding patterns and learn distinct interactions between proteins and ligands. This approach significantly improves the prediction accuracy for both seen and unseen ligands compared to existing methods.
2. The study addresses the limitations of traditional experimental methods and existing computational approaches by proposing a unified model that incorporates ligand information explicitly. LABind outperforms other advanced methods on multiple benchmark datasets, demonstrating its superior ability to generalize to new ligands and maintain robust performance even with predicted protein structures.
3. LABind’s architecture includes a graph converter module that encodes protein structures into graphs, capturing spatial features essential for binding site prediction. The cross-attention mechanism allows the model to effectively integrate ligand properties, enhancing its ability to distinguish between different ligands and their corresponding binding sites.
4. The application of LABind extends beyond binding site prediction to tasks like binding site center localization and molecular docking. It shows strong potential in improving docking accuracy and can be applied to proteins without experimentally determined structures by leveraging predicted structures from tools like ESMFold.
5. The study includes comprehensive experiments and ablation studies that validate the importance of each component in LABind, such as the protein representation and ligand features. The visualization of residue representations highlights how LABind captures crucial information about protein-ligand interactions, leading to more accurate predictions.
6. LABind demonstrates practical applicability by accurately predicting binding sites for the SARS-CoV-2 NSP3 macrodomain with unseen ligands, showcasing its potential in real-world scenarios. This method provides a valuable tool for understanding protein functions and aiding drug design efforts.
📜Paper: https://t.co/iIkeut8rNQ
💻Code: https://t.co/crButq4ykS
#ProteinLigandBinding #ComputationalBiology #MachineLearning #DrugDiscovery #Bioinformatics