The biological age of individual cell types can be evaluated using plasma proteomics, revealing diverse aging profiles across more than 40 cell types and links between the accelerated aging of specific cell types and disease.
https://t.co/cYxEDin5kc
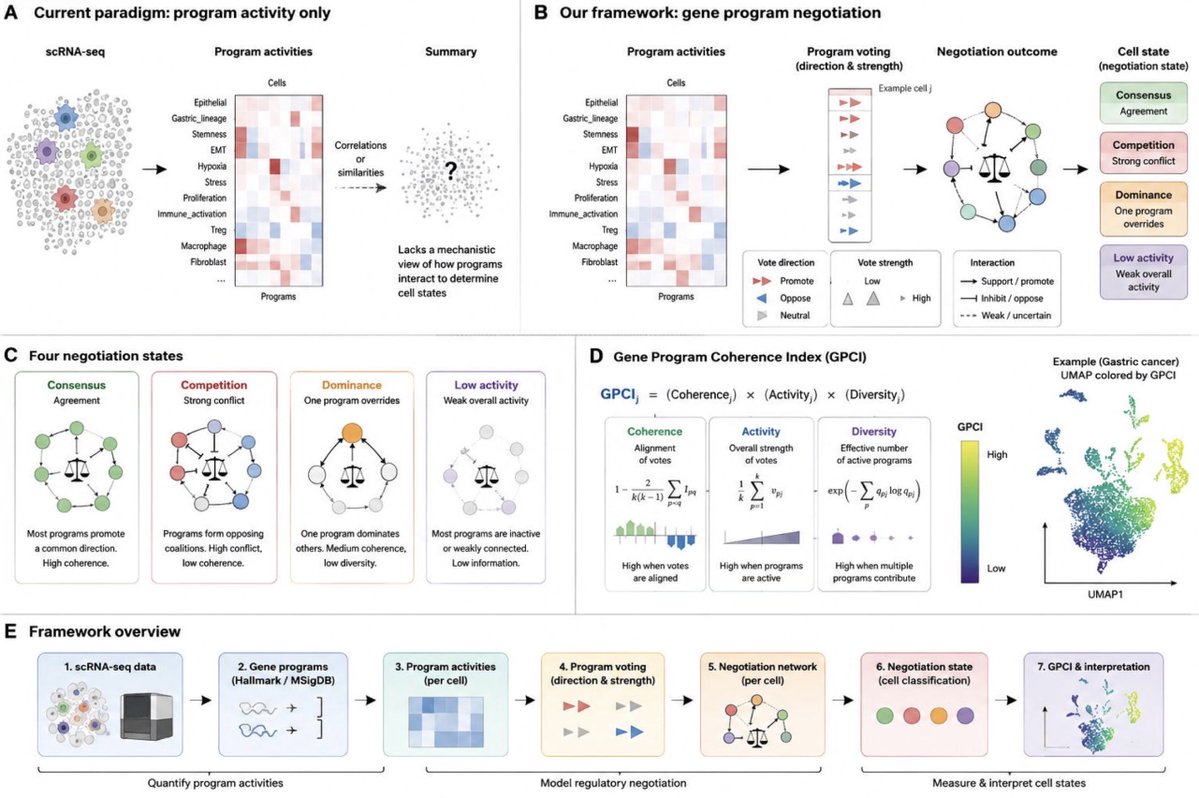
Gaining biological insights through supervised data visualization
If you have ever run t-SNE or UMAP on a biological dataset and gotten a beautiful plot that had nothing to do with the question you cared about, you have met the core problem this paper tackles.
Unsupervised embeddings preserve whatever variation dominates the data, which is often not the variation tied to your labels. The existing supervised fixes tend to overcorrect: they bake class membership into the distance metric and force artificially clean separation, which looks impressive but misleads you about the real structure. They also break on continuous labels and cannot place new unlabeled points.
Jake Rhodes and coauthors propose RF-PHATE, which threads the needle. Instead of distorting distances by class, they train a random forest on the labels and extract RF-GAP proximities, similarities defined by how often points land in the same terminal nodes, weighted to reproduce the forest's out-of-bag predictions. These proximities already encode feature importance for the supervised task.
They then feed them into a PHATE-style diffusion pipeline: row-normalize into a Markov operator, add a PageRank-like damping term so isolated clusters do not trap the random walk, power the operator for global structure, and map the potential distances to low dimensions with MDS. The result emphasizes label-relevant geometry while suppressing noise, and because random forests handle mixed and continuous targets natively, it works for classification and regression.
What makes the paper convincing is the stress-testing. On synthetic data with 500 added noise variables, RF-PHATE recovers the true branching structure while unsupervised methods collapse and class-conditional methods shatter it into fake clusters. They also introduce three metrics that penalize hyperseparation, then show across 27 datasets that RF-PHATE preserves structure without inflating separation, unlike supervised UMAP and S-tSNE. In multiple sclerosis data it surfaces a nonbenign RRMS subgroup, and on RNA-seq it holds cell-type separation even at 75 percent dropout where PHATE and UMAP fail.
The useful idea is that supervision can steer a visualization toward the variable you actually care about without manufacturing the separation you are trying to detect, the trap that makes most supervised embeddings useless for decision support. In drug discovery, clinical biomarker work, or materials screening, where you have noisy high-dimensional data plus a relevant label, this lets you explore structure that respects your target while staying honest about overlap.
Paper: Rhodes et al., Nature Computational Science (2026) , journal license | https://t.co/x1rrNJxnet
In our practice we often hear the same question. What should be done with exome sequencing data if no genetic diagnosis was found? About half of patients remain without an identified causal variant after the first analysis.🧬
A new paper in @NatureMedicine presents Talos which automatically reinterprets genomic data as new knowledge becomes available.
The tool identified 86 to 90 percent of known diagnoses in validation cohorts and performed similarly to Exomiser while returning only about one candidate variant per family for manual review.
Applied to 4735 previously unsolved cases Talos found 241 new diagnoses and increased diagnostic yield by 5.1%.
The older the original analysis the more new diagnoses became available. Additional yield reached 8.3% for cases analyzed in 2019.
The computational cost was surprisingly low. Monthly reanalysis of 1000 genomes costs only about $1.65 per year.
As AI and automated pipelines continue to evolve and partially take over repetitive interpretation tasks the answer seems increasingly obvious. Genomic analysis should not be a one time event. Every unsolved genome deserves periodic reanalysis.
https://t.co/dfiqoK5rxu
#ExomeSequencing #ACMG #Talos #InheritedDisorders
Large reasoning models as thinking machines for medicine
Most medical AI systems deployed so far are classifiers. You feed one a chest X-ray or a skin lesion and it returns a label with a confidence score. They're good at finding correlations, but they don't reason: they can't tell you why, ask a follow-up question, or reconcile lab values with patient history the way a clinician does.
The new generation of large reasoning models, the ones that articulate multi-step chains of thought rather than emitting a single output, opens a different door. In a recent perspective, Hong-Yu Zhou and coauthors map out what it would take to bring that capability into the clinic, under the name medical reasoning AI (MRAI).
The technical core is interesting for the ML community. Their proposed recipe layers three things on top of a pretrained transformer. First, post-training with reinforcement learning with verifiable rewards (RLVR): instead of training against live patients, the model explores reasoning paths inside a computational sandbox built from retrospective cases, and a checker scores each trajectory against ground-truth outcomes and clinical constraints. Critically, when a claim can't be verified from the available data, the reward engine abstains rather than reinforcing a guess, which curbs the spurious self-reinforcement that plagues naive RL.
Second, prompt optimization treated as a trainable component (think TextGrad-style textual backpropagation, DSPy pipelines) to cut the variance that makes raw prompting fragile in safety-critical settings. Third, agentic orchestration, where the model calls external tools (EHRs, PubMed, drug-interaction databases) and runs plan-act-verify-revise loops, with multi-agent setups mimicking a tumor board.
A useful part of the paper is the catalogue of what breaks. Reasoning traces are not faithful by default: a model can reach the right answer through wrong logic, and the chain it shows you may be a post-hoc rationalization rather than its actual computation. Verifying those traces adds clinician time, risks "verification fatigue," and demands evaluation of the reasoning process itself, not just final accuracy. Bias also moves from outputs into the reasoning steps, which is harder to detect.
The transferable lesson is the RLVR-in-a-sandbox pattern: if you can encode your domain's ground truth and hard constraints into a verifier, you can train reasoning models offline without risky live deployment. That's portable to drug discovery (lab-in-a-loop hypothesis refinement), materials development, and any pipeline where every intermediate step, not just the final prediction, has to survive expert scrutiny.
Paper: Zhou et al., Nature Biomedical Engineering (2026), journal license | https://t.co/MuAMMfTuUD
GenOT: generative optimal transport enables spatiotemporal interpolation and generation in cross-platform spatial transcriptomics https://t.co/v0T6Wskyq0
Learning to tune Ising machines on the fly: ML-driven combinatorial optimization for problems that never stop changing
Some optimization problems don't sit still. In wireless sensor networks, financial trading, or in-vehicle object tracking, you have to solve a fresh combinatorial problem every few milliseconds, where each new instance has a different size and structure and can't even be defined until the previous one is solved. Ising machines (specialized hardware that finds low-energy spin configurations) are fast at this, but they hide two practical traps: moving the large coupling matrix to the accelerator is slow, and the control parameters that give good solutions change with every problem. Searching for those parameters each time defeats the purpose.
Yohei Hamakawa and coauthors attack both problems at once. On the hardware side, they exploit a quiet structural fact: in many discrete problems, the coupling matrix takes only a handful of distinct values. They index those values and compute directly on the encoded form, shrinking data transfer and packing 4x more arithmetic units onto the FPGA. On the learning side, they train an XGBoost regressor on 2840 randomly generated graphs to predict the best simulated-bifurcation parameters straight from four graph features (node count, edge density, average degree, Gini coefficient). A second model picks whether the CPU or FPGA will be faster for each instance.
The interesting ML wrinkle is that the three control parameters aren't independent. Different combinations give identical solution quality, so learning them separately fails. They tie the targets to physically meaningful ratios instead, grounded in the bifurcation dynamics, which turns an ill-posed regression into a tractable one. The payoff: a maximum-independent-set solver up to 5500x faster than NetworkX, adapting its parameters and hardware choice per iteration with millisecond overhead.
What makes this matter for applied R&D is the shift from one-off tuning to learned, instance-aware configuration. Any pipeline that repeatedly solves shifting subproblems (route and resource allocation in logistics, real-time scheduling in manufacturing, candidate ranking in drug discovery, or grid dispatch in energy) can borrow this pattern: let a lightweight model set the solver's knobs so domain teams stop hand-tuning and start shipping. The hardware accelerator is the headline, but the reusable idea is treating parameter selection itself as a learning problem.
Paper: Hamakawa et al., Nature Communications (2026), CC BY-NC-ND 4.0 | https://t.co/t36zp7TAYJ
Sequence-based prediction of drug–target binding using machine learning, deep learning and ensemble models without 3D structural information
1. The study presents a fully sequence-based drug–target interaction (DTI) pipeline designed to work when reliable 3D protein structures are missing, aiming for docking-comparable discrimination while keeping the feature space interpretable.
2. Core idea: a unified, hand-engineered representation that concatenates (i) protein physicochemical descriptors (e.g., hydrophobicity/charge/composition), (ii) BLOSUM62-derived evolutionary information, (iii) protein 3-gram motif frequencies (local sequence context), and (iv) sequence-like drug encodings derived from canonical SMILES motif frequencies.
3. The framework is intentionally model-agnostic: it evaluates classical ML (Logistic Regression, SVM, Random Forest), deep learning (MLP, CNN), and multiple ensemble approaches (Extra Trees, Gradient Boosting, Histogram-based Gradient Boosting), plus a stacking ensemble.
4. The stacking classifier combines Random Forest + SVM + Logistic Regression via a meta-learner, leveraging complementary decision boundaries; reported mean ROC-AUC exceeds 0.90, with a maximum AUC of 0.914 under the paper’s protocol.
5. Evaluation emphasizes methodological hygiene: stratified 5-fold cross-validation, with fold-wise preprocessing to reduce leakage risk; scaling and SMOTE are applied only within training folds (validation/test folds remain untouched).
6. SMOTE’s impact is analyzed explicitly and shown to be model-dependent: it often improves minority-class sensitivity/recall, while effects on Accuracy/F1/ROC-AUC can vary across architectures—highlighting why imbalance handling must be reported alongside metrics.
7. Beyond scalar metrics, the paper inspects learning dynamics for deep models (CNN convergence behavior), confusion matrices for representative models, and ROC curves to characterize threshold-independent discrimination across folds.
8. Interpretability is treated as a first-class goal: permutation importance and SHAP analyses identify influential features, with protein-derived features (physicochemical properties and specific 3-gram motifs) frequently dominating—supporting biologically grounded explanations rather than opaque latent embeddings.
9. For orthogonal validation, the authors perform molecular docking (AutoDock Vina) on selected predicted pairs, using PDB structures and/or AlphaFold2 models filtered by confidence (pLDDT/PAE). A showcased case (EGFR) aligns high predicted binding probability with favorable docking scores (e.g., around −6.4/−6.2 kcal/mol), used as qualitative support.
10. Limitations are acknowledged: no independent external test set; stratified CV may still be optimistic if similar proteins/ligands appear across folds; docking validation is illustrative rather than a dataset-wide quantitative correlation—future work proposed includes stricter splits (cold-start/sequence-identity/scaffold splits) and broader docking benchmarks.
📜Paper: https://t.co/B9w1fArJ7D
#DrugDiscovery #ComputationalBiology #Bioinformatics #MachineLearning #DeepLearning #EnsembleLearning #DTI #Cheminformatics #ExplainableAI #VirtualScreening