Excited to share our work with @spence_jeffrey_ and @yun_s_song on detecting sample exchangeability in the absence of point labels!
https://t.co/1IGZyDwChK
Our work is a blend of stat theory, computation and application to real data, so I’ll try to explain each of these. 1/n
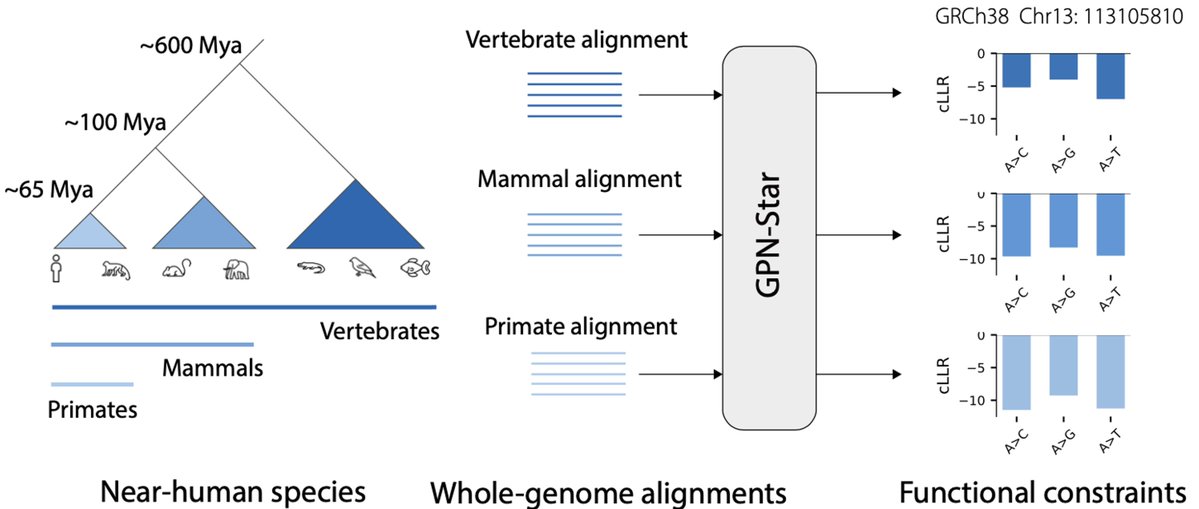
We are excited to share GPN-Star, a cost-effective, biologically grounded genomic language modeling framework that achieves state-of-the-art performance across a wide range of variant effect prediction tasks relevant to human genetics.
https://t.co/FTm3byYp67
(1/n)
We are grateful to be part of a kind and supportive scientific community. Discussions with Jessica Li (@jsb_ucla), Arjun Raj (@arjunrajlab), and members of the truly singular Song Lab (@yun_s_song) all helped shape this work. Thank you --- we'll pay it forward 🙏
Check out our work and software, led by the incredibly meticulous and capable @fanding_zhou! This is the culmination of work-in-progress presented a while ago at the 2022 Bioconductor Conference (see https://t.co/xo2HNuu414).
Gene expression changes aren’t just about mean shifts — variability shifts matter too, especially for aging. We're thrilled to introduce QRscore, a flexible non-parametric framework for detecting shifts in mean and variance across conditions. https://t.co/jfrK9JpJsE (1/7)
Thus with large n, we can hedge against the risk of model misspecification and maintain high statistical power. Even if we think that the distribution of the data is negative binomial. This theoretical insight underpins our method. #methodsmatter
Coincidentally, another article from my lab on DNA language models got published on the same day as GPN-MSA. It's freely available for 50 days from this link:
https://t.co/q8aWPIRGz8
Genomic language models: opportunities and challenges
Please share with your colleagues.
A DNA language model based on multispecies alignment predicts the effects of genome-wide variants @NatureBiotech
1. GPN-MSA, a DNA language model leveraging multispecies alignment (MSA), sets a new benchmark in predicting variant deleteriousness for both coding and noncoding regions of the human genome.
2. This model uses evolutionary insights from 100 vertebrate species to enhance predictions, outperforming existing DNA language models like Nucleotide Transformer and genome-wide predictors such as CADD.
3. GPN-MSA achieves superior results in classifying ClinVar, COSMIC, and OMIM variants, highlighting its utility in clinical and research contexts, including rare disease diagnosis and cancer genomics.
4. Unlike prior models, GPN-MSA is computationally efficient, requiring just 3.5 hours of training on 4 NVIDIA A100 GPUs while delivering high-accuracy predictions across 9 billion possible human variants.
5. It excels in distinguishing rare from common variants, leveraging conservation and genomic context, making it a robust tool for genome-wide variant effect prediction (VEP).
6. GPN-MSA's insights extend to functional impact predictions, capturing regulatory and epigenetic markers critical for gene expression and regulation studies.
7. The model paves the way for integrating DNA sequence modeling with functional genomics, offering potential breakthroughs in precision medicine, drug discovery, and population genetics.
@gsbenegas@cralbors@youngtableaux@chengzhong_ye@yun_s_song
💻Code: https://t.co/GNDhXAVbZw
📜Paper: https://t.co/ooNtfYUBmA
#GenomeVariants #DNAAnalysis #Bioinformatics #PrecisionMedicine #RareDisease #VariantPrediction
Happy New Year! Our GPN-MSA paper is finally published, under a slightly different title from the preprint. Please check it out and share it with your colleagues.
https://t.co/CKvTG2EZS2
1/4
Thrilled to present at #MLCB2023, about our latest work on #SpatialTranscriptomics - ggPair: A deep learning approach to unveil novel ligand-receptor interactions in cell-cell communication. 🧬
Catch my talk at 12:50 PM PT this Friday. All the talks are live-streamed on YouTube.
We recently posted a preprint describing GPN-MSA, a DNA language model that leverages whole-genome alignments across multiple species while taking only a few hours to train. This thread summarizes its performance on the human genome.
https://t.co/QyRBRqOXAX
1/12
After lengthy anticipation, we finally got to compare PrimateAI-3D with our Cross-Protein Transfer (CPT) model https://t.co/Sop1kUF8u6
Below is a summary of our findings, including a comparison of ESM-1b and ESM-1v protein language models.
1/8
Can current genomic sequence-to-expression models explain expression variation across individuals based on their personal genome? In the Ioannidis Lab at UC Berkeley, we evaluated 4 state-of-the-art models for this (Enformer, Basenji2, ExPecto, Xpresso)
https://t.co/WVpPMWQNcs
Interested in a fast, accurate method for estimating the parameters of a complex phylogenetic model of molecular evolution (e.g. a general rate matrix describing the co-evolution of protein contact sites in 3D)? If so, please check out our preprint: https://t.co/VnuGnBjByS
(1/10)
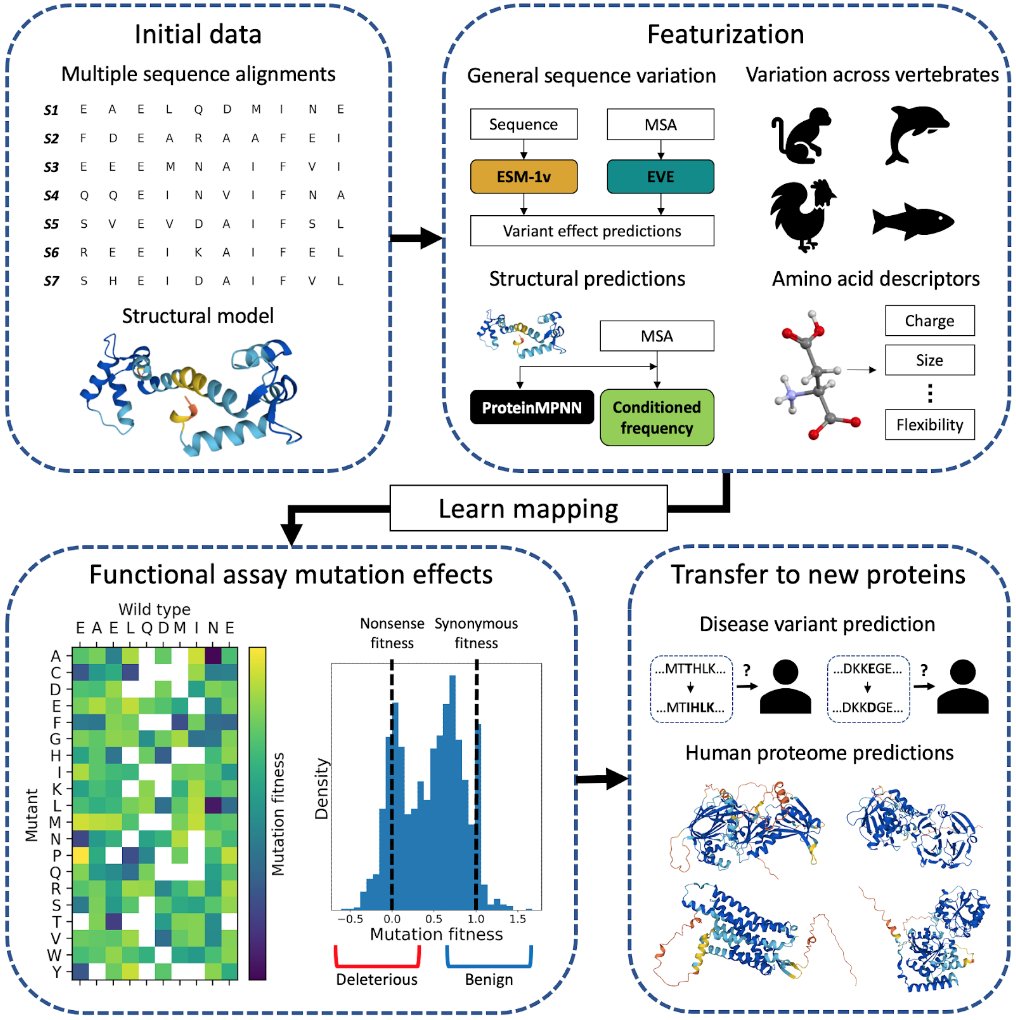
Predicting the effects of missense variants is a central problem in human genome interpretation. We are thrilled to share our preprint on using cross-protein transfer (CPT) learning to improve zero-shot prediction of disease variant effects:
https://t.co/Q4JLkLwNh8
(1/8)
Alan Aw @youngtableaux talked about the challenge of identifying marker genes in #scRNAseq and a new non parametric test for doing so
Work in progress 🏗 at https://t.co/gU6HQq8xpn
#BioC2022
Very glad to see this work by @gsbenegas published in eLife. https://t.co/Ryud4zq0Wi
We analyzed alternative splicing across diverse cell types in mice using scRNA-seq data from Tabula Muris and BICCN.
Many thanks to the reviewers for their thorough feedback.
The BAIR REU is back! @Berkeley_AI is currently recruiting undergraduates from HBCUs and PBIs for a hands-on summer 2022 research experience in artificial intelligence. More details about eligibility and support at https://t.co/JGy4kFF4Sx
Really excited to share a project I’ve been working on with Ryo Yamamoto and the Sudmant Lab. Uncovering tissue-specific changes in gene expression with age. We look at age-correlated gene expression and increasing expression heterogeneity with age. https://t.co/6pDBgtAwg8