For a detailed description + more experiments, check out our paper: https://t.co/ZSxjnmEM7N
Questions? Stop by our poster at ICML on Tuesday, July 7, 10:30am to 12:15 pm KST in Hall A (Poster #613)! (10/10)
Code: https://t.co/MvSGgxyigG
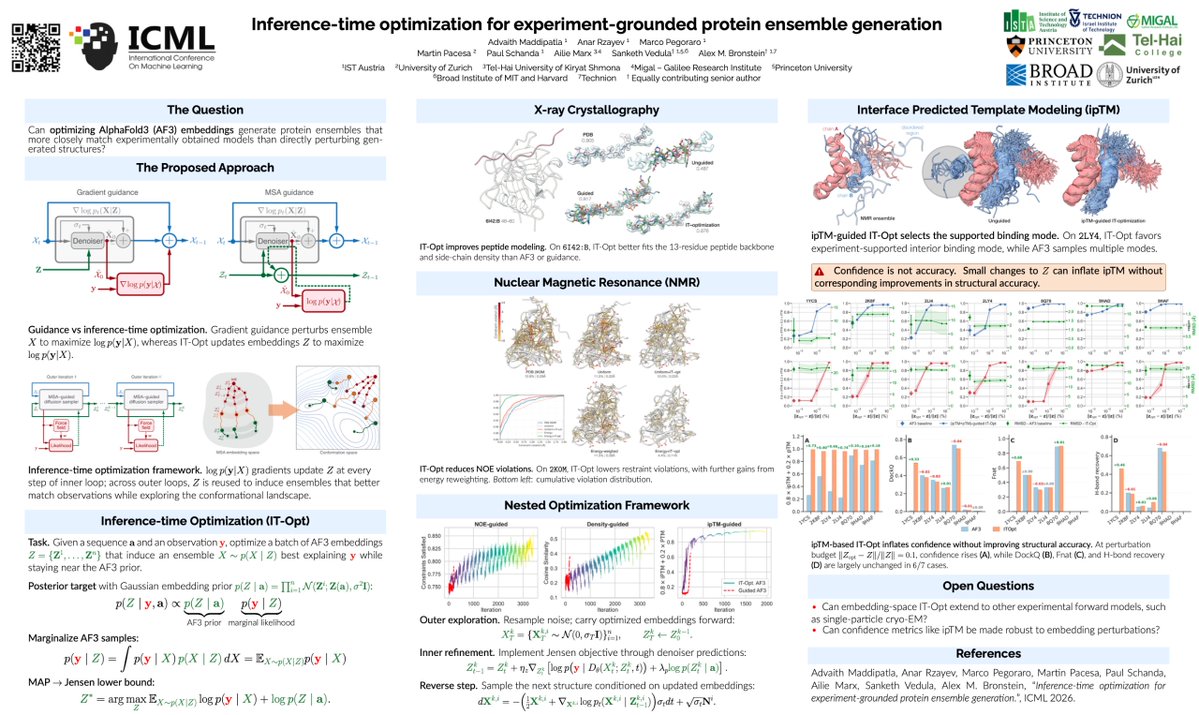
We also used IT-Opt to optimize surrogate objectives such as AF3 confidence scores. In some cases, this can improve binding-mode selection (left). But confidence ≠ accuracy: blindly optimizing these scores can inflate iPTM without improving structural agreement (right).⚠️(9/10)
Vision foundation models predict hurricane intensity from satellite imagery remarkably well — and pass out-of-distribution tests.
Yet their internal representations collapse exactly where physics matters most.
We call this the Perception–Physics Paradox. New at ICML 2026 🧵
Excited to be in Rio for ICLR 2026 🇧🇷
Tomorrow afternoon:
• HYPER: A Foundation Model for Inductive Link Prediction with Knowledge Hypergraphs
📍 Pavilion 3 — P3-#902
• Flock: A Knowledge Graph Foundation Model via Learning on Random Walks
📍 Pavilion 4 — P4-#5309
Inference-time Optimization for Experiment-grounded Protein Ensemble Generation
1. The paper introduces a novel inference-time optimization (IT-Optimization) framework that directly optimizes AlphaFold3's Pairformer embeddings to generate protein ensembles consistent with experimental data, fundamentally changing how experimental guidance is incorporated into structure prediction.
2. Unlike traditional gradient guidance that perturbs coordinates during reverse diffusion, this approach optimizes latent representations to maximize ensemble log-likelihood, eliminating dependence on diffusion length and removing initialization bias.
3. The framework employs a nested optimization structure with an outer loop for global exploration across diffusion trajectories and an inner loop for joint refinement of embeddings and structures, enabling persistent memory of experimental constraints across sampling runs.
4. The method introduces energy-weighted sampling schemes that combine AlphaFold3 structural priors with forcefield-based priors, allowing Boltzmann-weighted ensemble statistics rather than uniform weighting for thermodynamically plausible results.
5. On NMR benchmarks, IT-optimization substantially reduces NOE restraint violations compared to guided baselines, with further improvements when combined with energy reweighting using Amber99 force field.
6. For X-ray crystallography, the framework consistently outperforms experiment-guided AlphaFold3 across all evaluated benchmarks, achieving lower Rwork and Rfree values, improved local density alignment, and greater reproducibility across random seeds.
7. The method enables restraint-free modeling of protein-bound peptides and improves recovery of widely separated alternative conformations (altlocs), addressing key limitations of current approaches.
8. A critical finding reveals that interface predicted TM-score (ipTM) can be artificially inflated through very small perturbations of the embedding space (approximately 0.01%), exposing vulnerabilities in confidence metrics used for protein-binder design.
9. The ipTM optimization experiments demonstrate that increased confidence scores do not necessarily correlate with improved structural accuracy, suggesting cautious interpretation of such metrics in therapeutic development workflows.
10. The framework serves as a meta-guidance layer that composes with any modified sampling scheme, making it agnostic to internal diffusion iterations and applicable to diverse experimental modalities including NMR, crystallography, and potentially cryo-EM.
📜Paper: https://t.co/tRns09nJii
#AlphaFold3 #ProteinStructure #StructuralBiology #MachineLearning #ComputationalBiology #ProteinEnsembles #NMR #Crystallography #DiffusionModels #Bioinformatics
Experiment-guided AlphaFold3 resolves accurate protein ensembles
1. AlphaFold3 has revolutionized protein structure prediction by achieving near-experimental accuracy, but it often collapses to a single dominant conformation, missing the inherent heterogeneity of proteins. This new study shows how AlphaFold3 can be guided by experimental data to generate accurate ensembles of protein conformations.
2. The researchers adapted AlphaFold3 by incorporating experimental likelihoods into the reverse diffusion steps, allowing the model to generate ensembles that match data from NMR spectroscopy, X-ray crystallography, and cryo-EM. This approach significantly reduces distance restraint violations compared to traditional NMR structures.
3. When guided by NMR distance restraints and order parameters, AlphaFold3 produces ensembles with fewer constraint violations and better matches to experimental dynamics. This method also improves the accuracy of electron density fitting in X-ray crystallography, capturing previously unmodelled residues.
4. The study demonstrates that combining cryo-EM maps with NMR parameters further enhances model quality, providing a more accurate representation of protein complexes. This integration of modalities highlights the potential for experimentally aware predictive models.
5. The computational efficiency of this method, with runtimes of just a few minutes, makes it practical for large-scale applications. It could accelerate the analysis of legacy datasets and improve the interpretation of complex protein structures in various experimental contexts.
📜Paper: https://t.co/TWieuShG2H
#AlphaFold3 #ProteinStructure #ComputationalBiology #NMR #CryoEM #XrayCrystallography
Experiment-guided diffusion within AlphaFold3:
Inject differentiable likelihood gradients from experimental data into its reverse diffusion steps.
This steers sampling toward conformations that match real measurements.