AI can now read protein sequences directly from mass spectrometry data without relying on existing databases.
That could transform proteomics, biomarker discovery, and precision medicine.
Read more: https://t.co/ruzrAOUCSl #Proteomics#MachineLearning#ProteinSequencing #Bioinformatics #PrecisionMedicine #Biotechnology #MassSpectrometry
Foundation models come to mass spectrometry proteomics
Identifying proteins from their fragments is a foundational task in biology. A mass spectrometer breaks peptides into pieces, measures the masses, and software then tries to match each spectrum back to a peptide sequence. Two approaches have dominated for decades: database search (match against known proteomes) and de novo sequencing (infer the peptide directly from the spectrum). Both are bottlenecked by the same step, scoring how well a candidate peptide explains a spectrum.
Deep learning has been entering this field for years, mostly as feature extractors feeding traditional engines like MaxQuant or MSFragger.
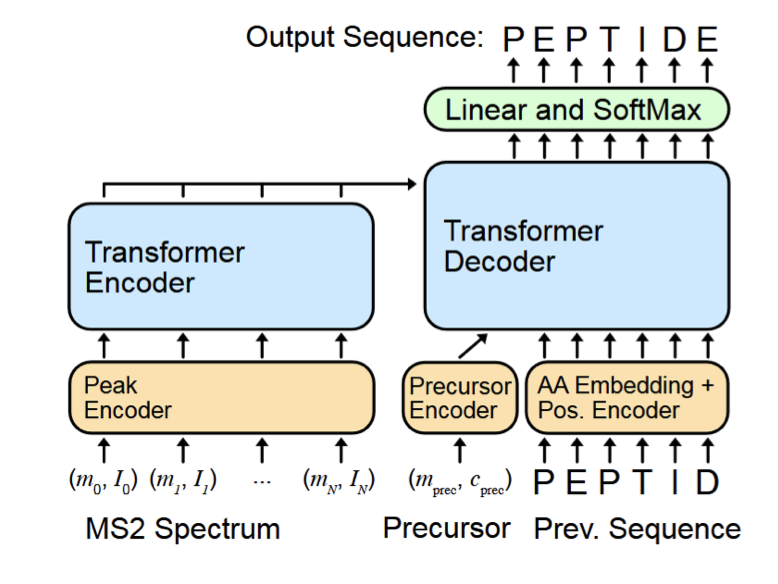
Jiale Zhao and coauthors introduce pUniFind, is a multimodal foundation model trained on over 100 million peptide-spectrum matches from open database searches. Spectra and peptides get their own encoders, and the model is pretrained with cross-modality tasks: predict the spectrum from the peptide, predict the peptide from the spectrum, score peptide-spectrum pairs jointly. Database search and de novo sequencing become two views of the same model.
The numbers are remarkable. In immunopeptidomics, where peptides come from non-tryptic digestion and are very hard to identify, pUniFind finds 42.6% more peptides than Open-pFind. In modification-rich de novo sequencing, it identifies 60% more peptide-spectrum matches than existing methods, despite working in a 300 times larger search space. In regular de novo, it recovers 38.5% more peptides, including 1,891 that map to the human genome but are absent from reference proteomes. A deep learning quality control filter raises consistency with RNA-Seq evidence from 65.4% to 85.0%.
What makes this an ML story is the architecture choice. End-to-end scoring with a shared latent space replaces hand-crafted feature pipelines, and the same backbone serves both database search and de novo sequencing without retraining.
For drug discovery, immunotherapy, and antibody engineering, peptide identification is the bottleneck for neoantigen discovery and biomarker pipelines. Tools that find more peptides at the same FDR, especially in non-tryptic and modification-rich settings, expand what is detectable from existing data. That changes what is worth running in the lab.
Paper: Zhao et al., Nature Machine Intelligence (2026) — journal license | https://t.co/U05n5GbjZR
@NielsRogge@arxiv Fantastic! Will the site automatically parse code repositories and HuggingFace datasets/spaces/models from the text or can we add those ourselves too?
@NielsRogge@ilyasut Would you be open to also include preprints published at bioRxiv @biorxivpreprint ? A lot of the AI for Science preprints are published there.
Diffusion Decoding for Peptide De Novo Sequencing
A new study investigates diffusion decoders for peptide de novo sequencing, addressing limitations of traditional autoregressive models like cascading errors and inefficient use of high-confidence regions.
The research demonstrates that while simply replacing a transformer decoder with a diffusion decoder initially lowered performance, incorporating the DINOISER loss function significantly improved amino acid recall by 0.373 compared to the Casanovo baseline.
This work highlights the potential of diffusion decoders to enhance model sensitivity and advance peptide de novo sequencing by allowing sequence generation to start from any peptide segment, leveraging high-confidence parts.
Despite the improvements in amino acid recall, the models with diffusion decoders still showed limitations in peptide precision and coverage, often producing sequences longer than the ground truth with noise at the end.
The study used Casanovo as a development framework due to its relatively lower computational demands and higher performance, training and evaluating models on a combined tryptic and non-tryptic dataset.
The authors explored three different diffusion decoder designs (Casanovo-DS, Casanovo-DM1, and Casanovo-DM2) and evaluated various loss functions, finding DINOISER to be optimal for amino acid recall.
Knapsack beam search was also investigated but resulted in worse metric performance and significantly increased training time, from approximately 1.5 hours to 35 hours for the tested models.
Future work aims to address the issue of excess noise in predicted sequences, explore hyperparameter tuning for diffusion decoder models, and conduct fair comparisons with other state-of-the-art methods using consistent datasets.
📜Paper: https://t.co/b7AbuWOIxj
#PeptideSequencing #DeepLearning #DiffusionModels #MassSpectrometry #Bioinformatics
Pairwise Attention: Leveraging Mass Differences to Enhance De Novo Sequencing of Mass Spectra
1. This study introduces Pairwise Attention (PA), a transformer-based model that explicitly encodes pairwise mass differences between spectrum peaks as a learned bias, significantly improving de novo peptide sequencing accuracy.
2. Inspired by how experts manually interpret MS2 spectra—by detecting consistent mass differences between peaks—PA integrates this inductive bias directly into the attention mechanism of the transformer encoder.
3. PA adds only 0.1% more parameters to the model while achieving a 14.2% (6.7 percentage point) improvement in peptide-level precision at 100% coverage over a baseline transformer, and a 7.2% (3.6 point) gain over the state-of-the-art Casanovo.
4. The learned pairwise mass difference bias allows the model to better capture fragmentation patterns like b- and y-ions, enhancing interpretability and boosting accuracy on both clean and noisy spectra.
5. Evaluation on the nine-species benchmark (versions V1 and V2) shows consistent gains for PA across all species, outperforming Casanovo on most tasks, especially for species with fewer spectra or lower data quality.
6. On an independent bacterial dataset, PA generalized better than both the base model and Casanovo, achieving an 8.5 percentage point precision gain—demonstrating robustness beyond benchmark data.
7. Despite the added memory and training time cost (~31% slower), PA’s improvements are robust across datasets, model configurations, and random seeds, suggesting the utility of domain-specific feature engineering in deep learning.
8. The PA model architecture retains the standard decoder from Casanovo but modifies the encoder with a Fourier-encoded pairwise m/z difference matrix, injected into the self-attention layers as a learnable bias.
9. Results show that the base transformer may struggle to fully learn pairwise spectral features from scratch, whereas explicitly encoding this structure improves convergence and final performance.
10. The authors provide an open-source implementation of the PA model, enabling integration with other transformer-based mass spec models, and setting a new standard in interpretable, domain-aware de novo sequencing.
💻Code: https://t.co/Az60fWnTxl
📜Paper: https://t.co/Ar6xwhmBuH
#MassSpectrometry #DeNovoSequencing #Proteomics #TransformerModels #DeepLearning #Bioinformatics #MS2 #PeptideIdentification #AttentionMechanisms #ComputationalBiology
AbNovoBench: A Resource and Benchmarking Platform for Monoclonal Antibody De Novo Sequencing
1 The field of monoclonal antibody de novo sequencing finally has a comprehensive benchmarking platform it desperately needed. AbNovoBench introduces the largest high-quality dataset to date with over 1.6 million peptide-spectrum matches from 131 antibodies across six species and 11 proteases.
2 What makes this resource truly valuable is the systematic evaluation of 13 deep learning-based de novo peptide sequencing algorithms using unified training data and standardized metrics, enabling fair comparison across methods that was previously impossible due to inconsistent evaluation protocols.
3 The study reveals that transformer-based architectures, particularly ContraNovo and Casanovo variants, consistently outperform recurrent neural network and graph-based models across amino acid, peptide, and post-translational modification accuracy metrics.
4 A surprising finding challenges the assumption that bigger training datasets always yield better results. Casanovo v1, trained on just 2 million PSMs with three common modifications, achieved comparable or superior PTM precision to ContraNovo trained on 30 million PSMs, suggesting domain-informed constraint can be more valuable than raw data volume.
5 The platform introduces antibody-specific evaluation metrics including coverage depth analysis for complementarity-determining regions, which are critical for assembly but often underrepresented in sequencing outputs compared to constant regions.
6 For full-length antibody reconstruction, the template-guided Fusion assembler achieved error-free reconstruction when combined with high-quality peptide predictions, while fully de novo assembly remains challenging for unknown sequences.
7 The analysis identifies key limiting factors affecting all algorithms: peptide recall drops sharply beyond 19-20 residues, missed cleavage sites significantly impair performance, and spectral noise shows strong negative correlation with accuracy across all models.
8 Cross-enzyme robustness testing reveals substantial performance variations, with transformer models maintaining stability across both tryptic and non-tryptic conditions while other architectures degrade significantly under challenging cleavage regimes like thermolysin and Asp-N.
9 The web platform at https://t.co/yHjFbDFYZo provides pre-trained models, curated datasets, and customizable workflows, eliminating the need for researchers to retrain models from scratch and enabling real-time tracking of methodological advances in the field.
10 The study acknowledges current limitations including reliance on HCD fragmentation data and the need for expansion to EThcD, UVPD, and data-independent acquisition workflows in future platform updates.
💻Code: https://t.co/NgLLqffsbO
📜Paper: https://t.co/B1G9DqOmsK
#AbNovoBench #MonoclonalAntibody #DeNovoSequencing #MassSpectrometry #Proteomics #DeepLearning #Benchmarking #Bioinformatics #ComputationalBiology #AntibodyEngineering
π-MSNet provides an AI-ready data framework for efficient training and systematic benchmarking of multiple models across three representative tasks (e.g., MS/MS spectrum prediction, retention time prediction, and de novo peptide sequencing).
The AI models, called InstaNovo and InstaNovo+, are a step toward “the holy grail” of protein research: to unravel the genetic identity of previously unstudied proteins en masse https://t.co/eiozqndarU
AI has been particularly groundbreaking in helping scientists understand protein structures — the workhorses of living cells. The #technology has now taken a massive step forward with #InstaNovo, a new tool designed to advance protein sequencing. https://t.co/A3cyBZUNvW
#AI#daily
InstaNovo+, launched July 4, 2025, uses diffusion AI to sequence peptides, advancing proteomics and drug discovery with high accuracy.
Geo impact: worldwide
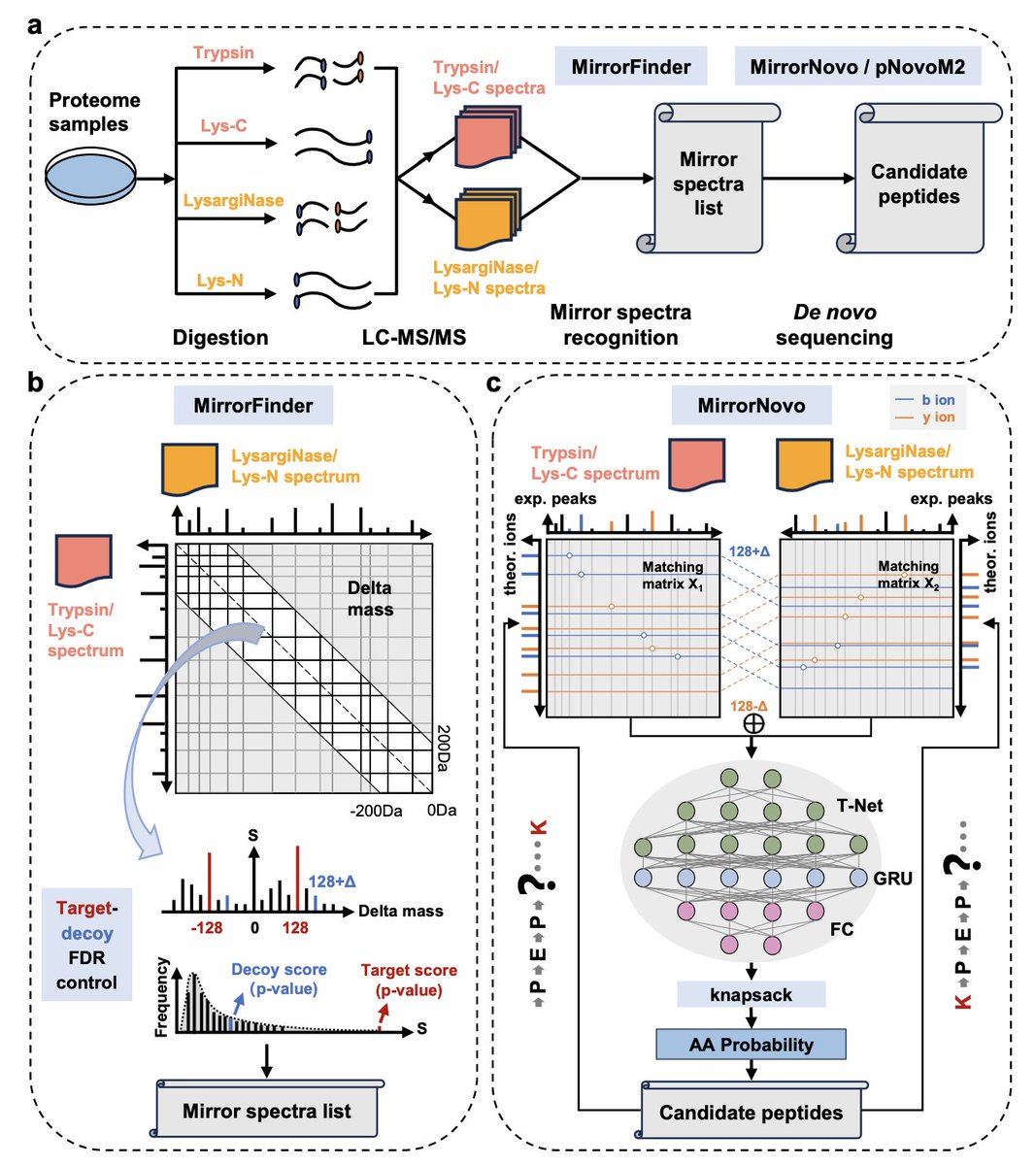
Combining mirror proteases with deep learning sets a new ceiling for de novo peptide sequencing
Mass spectrometry-based proteomics depends on breaking proteins into peptides before sequencing them. The problem: no single enzyme does this cleanly. Trypsin—the workhorse of proteomics—leaves many peptides undigested, and incomplete fragmentation means critical sequence information simply goes missing. The result is systematic blind spots in what we can identify.
Mirror proteases offer a physical solution. Trypsin cleaves C-terminal to lysine and arginine; LysargiNase cleaves N-terminal to the same residues. The resulting peptide pairs share an identical internal sequence but carry the cleavage site at opposite ends—and their mass spectra are complementary. Fragment ions absent in one are present in the other.
DiNovo (by Cao and coauthors) builds a full computational system around this idea. The core sequencing engine, MirrorNovo, is a deep neural network that takes both spectra of a mirror pair as simultaneous input. A T-Net extracts peak features from two matching matrices—one per spectrum—aligned at their shared fragmentation sites. A GRU layer captures dependencies across peaks, and fully connected layers output amino acid probabilities constrained by a knapsack algorithm. The system runs two pairs of mirror proteases (trypsin/LysargiNase and Lys-C/Lys-N) in parallel, then merges results.
A companion algorithm, MirrorFinder, identifies mirror spectral pairs without pre-sequencing—using precursor mass differences and fragment ion distributions directly, with target-decoy FDR control. A third innovation, TD mapping, evaluates de novo results against a reference database at matched FDR, enabling the first direct performance comparison between de novo sequencing and database search.
The numbers are remarkable. Mirror spectra reach 98% average fragment ion coverage versus 90% for single-protease spectra. Compared to trypsin alone, DiNovo sequences 154–195% more high-confidence amino acids and identifies 29–34% more proteins. Across all tested algorithms (pNovo3, PointNovo, GraphNovo, Casanovo), DiNovo sequences 110–195% more high-confidence peptides with 36–74% higher amino acid coverage. And it identifies a comparable number of proteins to established database search engines.
Sequencing proteins from species with no reference genome, characterizing neoantigens, reading antibody sequences: all require moving beyond the limits of single-enzyme experiments. DiNovo shows that pairing the right experimental design with a neural network built for it substantially expands what proteomics can reach.
Paper: https://t.co/16BHzWzceB
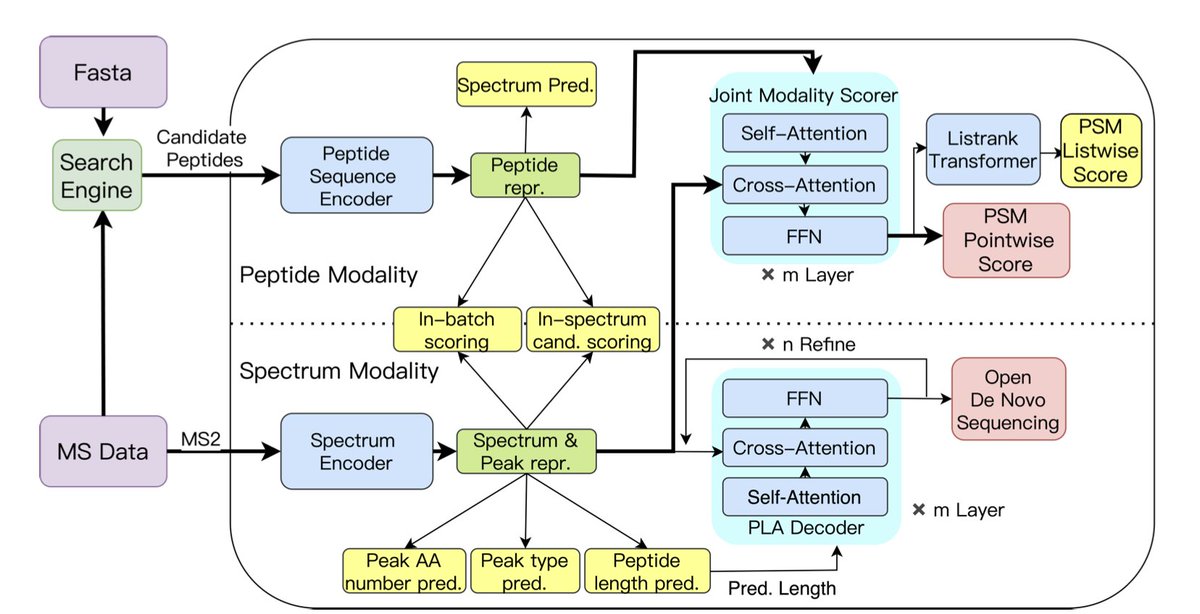
pUniFind: a unified large pre-trained deep learning model pushing the limit of mass spectra interpretation
1.pUniFind introduces the first large-scale multimodal pre-trained deep learning model in proteomics that unifies end-to-end PSM scoring and open de novo peptide sequencing. It’s trained on 100M+ spectra annotated via open search, enabling broad modification support and zero-shot capability.
2.In immunopeptidomics, pUniFind identifies 42.6% more peptides than traditional engines and achieves a 60% increase in PSMs over existing de novo methods, despite a 300-fold larger search space. It also discovers peptides absent from known proteomes but present in the genome.
3.A novel deep learning–based quality control strategy improves result reliability by filtering low-confidence spectra, recovering 38.5% more peptides with full fragment ion coverage. Among them, 1,891 were found only in the genome-translated database, highlighting pUniFind's discovery power.
4.Unlike most existing models, pUniFind supports over 1,300 peptide modifications and performs open de novo sequencing with no prior knowledge. It outperforms traditional tools like pNovo across 21 PTM datasets, achieving a 63.8% recall vs. pNovo’s 47.6%–39.8%.
5.Cross-modality pretraining tasks—de novo spectrum prediction and peptide reconstruction—improve spectrum-peptide alignment. This enables robust generalization across instruments and species, validated through mixed-species entrapment and metabolically labeled datasets.
6.pUniFind achieves consistent performance improvements across nine species and multiple instruments (QE, timsTOF, Astral), and remains accurate under large-scale or modification-heavy conditions like metaproteomics and immunopeptidomics.
7.In head-to-head comparisons, pUniFind surpasses traditional and modern tools such as Open-pFind, MSFragger+MSBooster, MaxQuant, Casanovo V2, DDA-BERT, and Tesorai, both in identification yield and in controlling false discovery rates.
8.A two-mode open de novo workflow (regular and modification-enriched) allows pUniFind to adapt flexibly to both general and PTM-enriched datasets. It uses pFind as a second-pass scorer to refine predictions for rare modifications.
9.De novo predictions include peptide length estimation and modification token prediction. Modifications are encoded by type, not site, to better leverage similar-mass modifications and enable site-independent pretraining.
10.On real-world S. cerevisiae data, pUniFind's open de novo sequencing recovers 40.2% more peptides than Casanovo V2. Its top 6 identified modifications match those reported by Open-pFind, confirming modification discovery robustness.
11.In immunopeptidomics, pUniFind achieves 86.3% peptide recall in de novo mode—significantly higher than Casanovo V2 (69.1%). Structural modeling confirms that novel peptides predicted by pUniFind bind reliably to HLA molecules, with AlphaFold3 ipTM scores above 0.90.
12.Inference is efficient and scalable: analyzing 24 raw files takes ~10 minutes on 8×4090 GPUs, compared to Open-pFind’s 5-hour open search. This makes pUniFind practical for high-throughput proteomics pipelines.
13.Future directions include incorporating retention time into scoring, adapting the model for DIA data, and further expanding the pretraining corpus to enhance rare-modification coverage and non-tryptic digestion modeling.
💻Code: https://t.co/nLnvI5A80e
📜Paper: https://t.co/DeHylSsOA1
#proteomics #deeplearning #massspectrometry #peptidesequencing #computationalbiology #HLA #PTM #OpenSearch #DeNovoSequencing
Discover DiNovo! Achieve high-coverage, high-confidence de novo peptide sequencing with mirror proteases & deep learning. Revolutionizing protein digestion! PMID:41786727, Nat Commun 2026, @NatureComms https://t.co/derMWdgLNg #AI#Pharma#BioMed#RNA#ASHG#ESHG