I am very happy to announce that we solved the in-virus #cryoEM structure of the #HIV matrix protein. The structure helped us to resolve a long mystery in HIV biology. A🧵1/5
Read more in @Nature : https://t.co/rVDC5LomAn 🧪
Mature #HIV-1 contains >2000 copies of “spacer peptide 2”, but we didn’t know why. We now see that SP2 binds the matrix protein and triggers the change into its mature arrangement. Summary video from @MargotRiggi :
New essay: A Brief History of Bioinformatics Software
Although the word "bioinformatics" wasn't coined until 1970, the first computer program to analyze protein sequences, named COMPROTEIN, was published in 1962.
From our forthcoming book, "Making the Modern Laboratory."
I think this is one of the most important articles we've published at @AsimovPress. If you read carefully, there are at least 3-4 ideas in here that *should* be large, well-funded research programs.
The article begins by arguing that existing AI models are good at predicting things *within* an existing framework, but are not good at building new frameworks (and, thus, cannot do paradigm-shifting science). As AI models become more widespread in science, they therefore risk "hypernormal science," meaning we will have less actual breakthroughs and more incremental discoveries.
The author (Alvin Djajadikerta) supports this argument with several examples, one of which comes from germ theory:
"In the mid-nineteenth century, doctors thought that illness was caused by noxious air, and kept meticulous records accordingly. The physician William Farr mapped cholera deaths across London and found they correlated strongly with low elevation, which he thought was because noxious vapors accumulated in low-lying areas. He was actually picking up a real signal: low-lying districts were closer to the contaminated Thames River. But because his data was organized around air quality, he could not find the true cause..."
"An AI trained on Farr’s records could have found even subtler correlations, and would have been genuinely useful for predicting which neighborhoods would be hit hardest in the next outbreak. But it would not be able to derive the concept of a waterborne microorganism, as this was not a variable anyone had yet recorded."
After giving other examples of this, Alvin begins mapping out ideas to solve this problem and create AIs that are "visionary" rather than "merely predictive." My favorite idea, of his, is to use AI agents as a model organism for metascience.
The gist is that many paradigm shifts seem to happen under particular conditions. "Bell Labs, Xerox PARC, and the early Laboratory of Molecular Biology at Cambridge all produced extraordinary concentrations of paradigm-shifting work," Alvin writes, "mostly because they were small groups with enough institutional protection to pursue ideas that looked unproductive by conventional measures."
Alvin continues:
"We have never been able to run controlled experiments on scientific institutions; it is impossible to create labs that differ in only one respect and compare the results. But we could run AI agents in parallel populations under different research conditions, and analyze the results...In this sense, AI scientists may give metascience its first model organism."
"For instance, one could test how group structure shapes discovery: do small, isolated teams produce more conceptual reorganization than large, well-connected ones? Do flat hierarchies outperform rigid ones? One could run AI agent populations that vary these factors independently and measure the results — something that is impractical to do with real institutions..."
This essay is excellent throughout and I hope you'll read it.
Researchers killed Mycoplasma capricolum cells, confirmed they were dead, and then "revived" them by transplanting in a synthetic Mycoplasma mycoides genome.
I'll plan to write a longer essay on this.
Very happy to see this work from Fabian Rehm, Jason Chin, and team on the development of a high error-rate orthogonal DNA replication system in E. coli, thus supporting continuous hypermutation and evolution of target genes in vivo. The system is based on a protein-primed linear plasmid replication mechanism, which has become a reliable way of realizing the orthogonal replication concept through which hypermutation is durably targeted to an orthogonal plasmid while sparing the genome. Our original orthogonal DNA replication (OrthoRep) system in yeast, Rongzhen Tian's and Jason Chin's BacORep and EcORep systems in bacteria, and now this promising new system all repurpose protein-primed replication to achieve orthogonality. I look forward to seeing how these systems continue to be applied to gene and biomolecular evolution at scale! https://t.co/HYLmVPOftV
We just made an app that walks you through designing a novel protein with AI from scratch. Takes about 5 minutes, requires zero biology knowledge.
➡️ https://t.co/L3dg6H6BTU
The best part: we will actually synthesize 1000 of those protein designs in the lab and test their real world function as a therapeutic.
IT'S HAPPENING! 💥 I'm psyched to launch the collaboration between @qedScience & @openrxiv@biorxivpreprint! Let's go back to concentrating on making discoveries, and not on “getting published”, because it’s not going to matter soon, when feedback & evaluation are detached from the journals. Preprint + q.e.d = your science is out there, and anyone can appreciate it 👇
Group Leader in Biomedical Engineering at the
@MDC_Berlin in Berlin.
It's a broad call for anyone doing exciting bioengineering; please apply or share. We have a great institute with amazing colleagues and core facilities,
highly recommend!
https://t.co/XB7voIyBJE
Maximum Likelihood seems like such a natural idea, but it has historically been highly controversial, with an epic and turbulent history with numerous assaults on the idea, culminating in a beautiful and complicated theory.
A highly entertaining read:
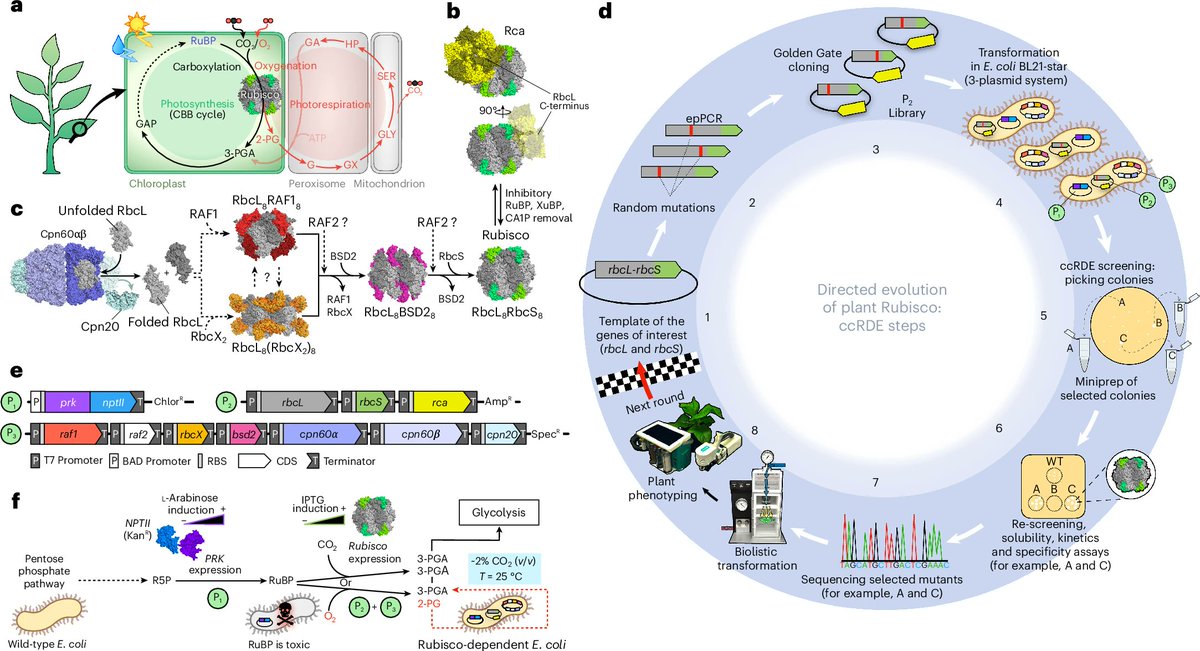
Rubisco is (arguably) the most abundant protein on Earth. (LPP surely comes close, right?) It’s an enzyme that fixes CO₂ into sugars during photosynthesis.
Unfortunately, as most people learn in school, Rubisco is inefficient. Sometimes it confuses O₂ for CO₂ and wastes energy. Plants make up for this in raw concentration; up to half the soluble protein in a leaf is Rubisco.
People have been trying to engineer better Rubiscos for many decades, but it's not easy because the proteins are big, do not fold easily (they need chaperone proteins to help out), are made from 16 subunits in land plants.
But there's a new paper in Nature Plants that looks really interesting. The TL;DR is that a group in Australia figured out how to express plant Rubiscos (and all SEVEN of their folding chaperones) using a set of 3 plasmids inside of E. coli cells. This enabled them to do "directed evolution" of Rubisco in bacterial cells, and quickly find Rubisco mutants that have higher enzymatic efficiency or that fold better.
In addition to the 3 plasmids, the researchers also coaxed E. coli to make ribulose-1,5-biphosphate, or RuBP, which is the 5-carbon sugar that Rubisco smashes into carbon dioxide to make molecules of 3-PGA for central metabolism.
Now, the clever bit is that you RANDOMLY MUTATE the three plasmids encoding the Rubisco to make millions of variants. Then, you transform those mutated plasmids into E. coli. If the E. coli do NOT make a functional Rubisco, RuBP levels build up and kill the cell; the molecule becomes toxic. But if the E. coli DO make a functional Rubisco, then they keep the RuBP levels in check and live just fine.
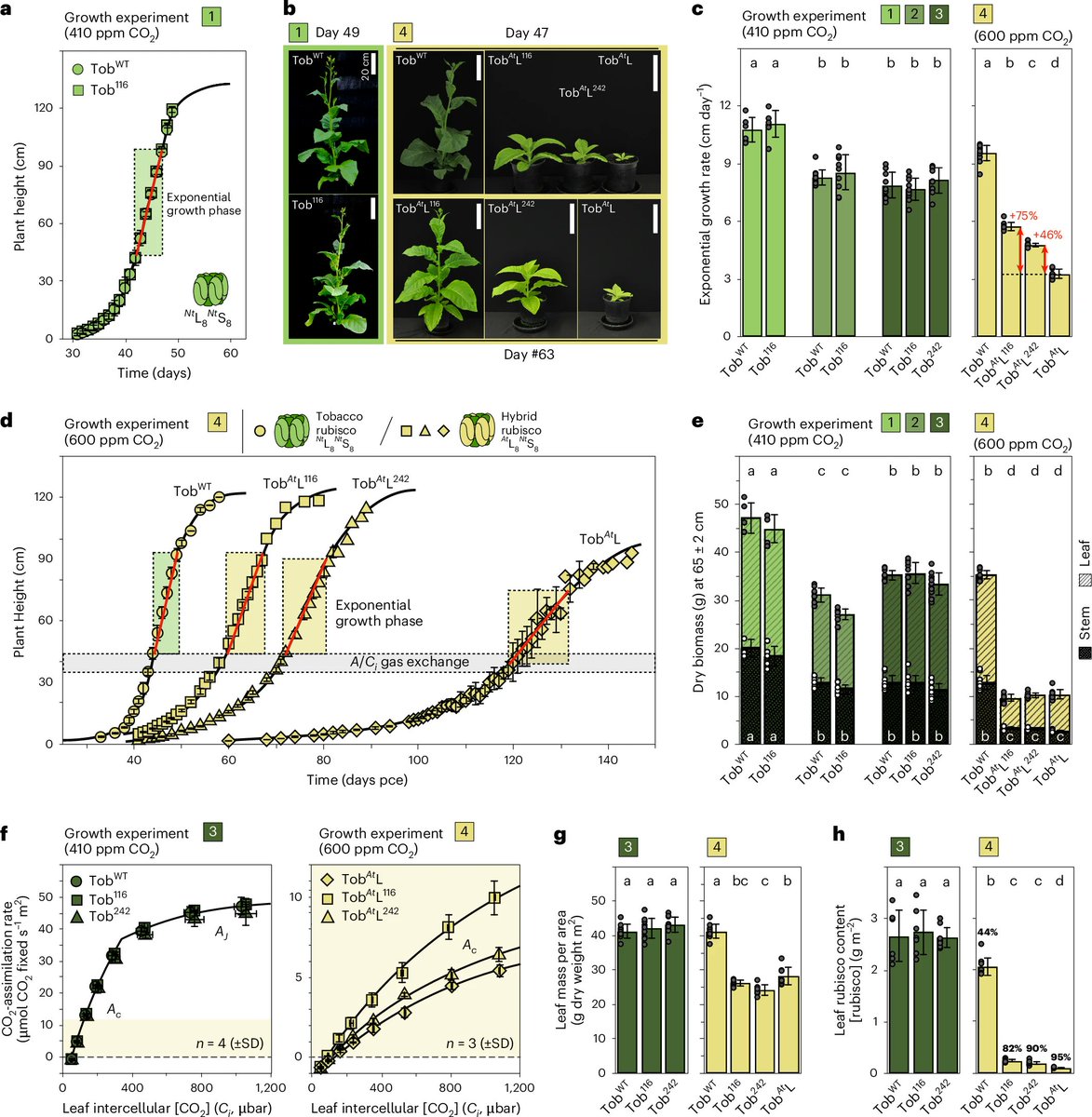
Using this "screening assay," the researchers found 46 fast-growing colonies of E. coli. Two of those colonies encoded really useful mutations. One mutation (M116L) makes Rubisco about 25–40% faster. The other (A242V) makes it fold and assemble much more efficiently.
They put this mutation into a "hybrid Arabidopsis–tobacco Rubisco," put that into tobacco plants, and measured growth. The plants with M116L grew 75% faster than wildtype.
No guarantees this will scale to more useful crops, like wheat and corn and soybeans etc. But it seems like a nice in vitro assay for faster prototyping!
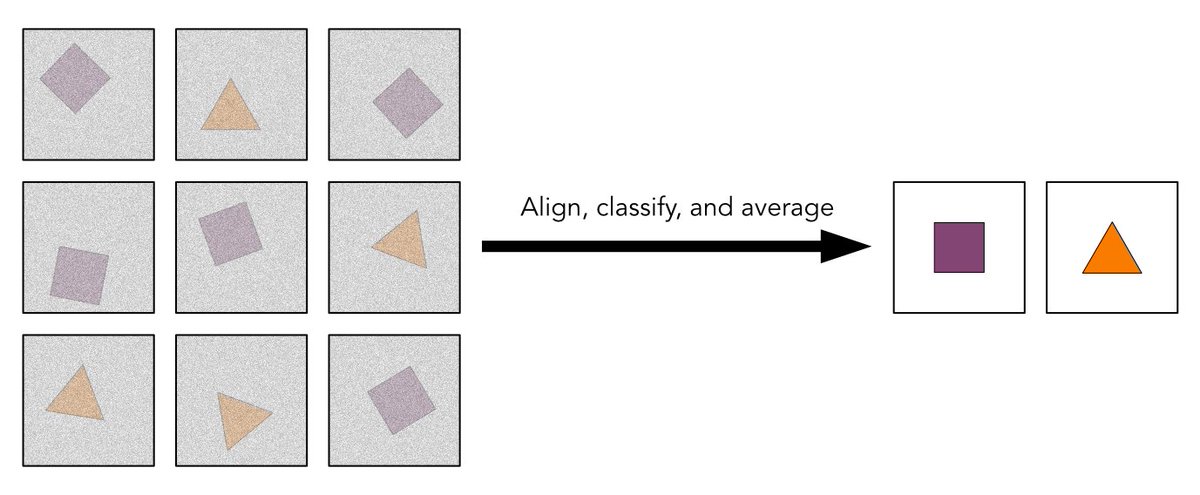
Looking for a deep dive into 2D classification in #cryoEM?
These recent guide pages cover practical aspects & parameter choices when running these jobs in #CryoSPARC, along with the theory behind expectation maximization: https://t.co/vpWLIlrBK3 and https://t.co/TKUYlvhruy
CodonTransformer: a multispecies codon optimizer using context-aware neural networks
Scientists looking to produce proteins in new host organisms often face challenges in adjusting DNA sequences to match the host’s translation preferences. This customization, known as codon optimization, can improve expression of both natural and engineered proteins in bacteria, yeast, plants, and mammals. However, designing sequences that preserve long-range dependencies and avoid regulatory pitfalls requires sophisticated methods and extensive data.
Fallahpour et al. developed a Transformer-based deep learning model, trained on over one million gene-protein pairs spanning 164 species, to address these complexities. Their architecture applies a BigBird Transformer variant with block-sparse attention, incorporating specialized tokens that represent both amino acids and codons. Through a masked language modeling strategy, the authors introduced a framework called STREAM that aligns codon and amino acid information, enabling the model to generate complete DNA sequences from partial inputs. They further fine-tuned the model using the top 10% of genes with the highest codon similarity index and demonstrated context-specific optimizations that captured organism-level preferences.
The researchers found that their approach produces DNA sequences with a near-natural balance of rare and common codons, matching local frequency distributions and maintaining GC content. They also reported minimal disruption by unwanted cis-regulatory elements, making these outputs potentially suitable for high-yield protein production. This combination of accurate language modeling, comprehensive training data, and flexible fine-tuning illustrates the model’s potential for robust codon optimization across multiple species.
Paper: https://t.co/a4nbZTMZbf
Introducing ATOMICA 💫
A model to universally represent molecular interactions (for proteins, nucleic acids, small molecules, and ions) at an all-atom scale 🧵