Prof. of Digital Innovation @UniMelb. Computational Scientist & Chemist, HPC & AI engineer, co-founder at QDX. Enabling the Digital Chemistry Revolution.
So far there is no evidence that an algorithm using a QPU can scale to any biosystem, let alone run ensemble statistics (e.g. via MD). Considering the latency of current QPUs, it would be technically impossible. I agree is fantastic that people are trying, and there is huge potential in the QC on QPU field (I definitely would not start with biosystems though!). However, over claiming with no supporting evidence is only damaging good research.
To this date and time there is sufficient evidence, or lack of thereof, to safely state that "Current quantum computing algorithms fail to describe quantum systems accurately and efficiently enough to be of practical use in drug discovery"
Honoured to receive the 2025 Dirac Medal from @WATOC. This is a shared achievement with my teams at @unimelb and @ANUComputing, our collaborators, and mentors—thank you all. More here: https://t.co/XoFr0KZ8Uo
@CompChemBioBot
An eight-member team has received the 2024 ACM #GordonBellPrize for their project, "Breaking the Million-Electron and 1 EFLOP/s Barriers: Biomolecular-Scale Ab Initio Molecular Dynamics Using MP2 Potentials."
Paper: https://t.co/9OHabtEREO
Learn more: https://t.co/7Pn0EQx7kT
🏆 Huge congratulations to a team of Frontier users led by @UniMelb for winning the 2024 Gordon Bell Prize for their groundbreaking #quantum molecular dynamics simulation! 🎉
Congratulations to all the winners today at @Supercomputing#SC24!
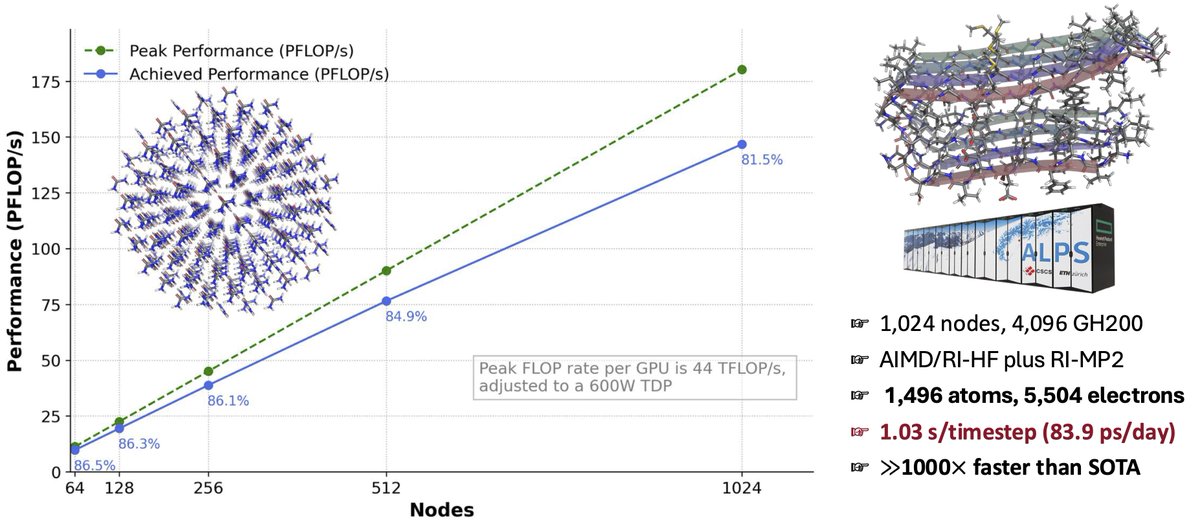
Sharing some initial results from our recent runs of Ab Initio Molecular Dynamics (AIMD) using EXESS on the ALPS (@cscsch ) supercomputer, pushing up to 4096 GH200 @nvidia superchips.
All calculations were performed in double-precision (FP64) at the AIMD/MBE3/RI-HF plus RI-MP2/cc-pVDZ (quantum) level of theory. This was applied to the whole system, no QM/MM or similar.
The left image demonstrates strong scaling efficiency, achieving 81.5% of FP64 R-Peak across 4096 GH200.
On the right, we highlight the 2BEG system—an amyloid fibril model that we are working on as relevant to Alzheimer’s research, comprising 1496 atoms and 5504 electrons.
With 4096 GH200, we reached on 2BEG a timestep latency of 1.03 seconds, translating to over 83.9 ps/day, safely marking a >1000x acceleration over the state-of-the-art at this level of theory.
Join us at @Supercomputing for our #GordonBellPrize talk to dive deeper into our work: https://t.co/Z44mHLkB2l
#HPC #quantumchemistry #drugdiscovery #GPU
#compchem #SC24
The cat is out of the bag! Our team is officially one of the six finalists for the @TheOfficialACM 2024 #GordonBellPrize—the highest award in high-performance computing. 🏆 Looking forward to discussing more about what we have achieved at @Supercomputing 2024 in November!
Read the SC announcement here: https://t.co/879mP39zi7
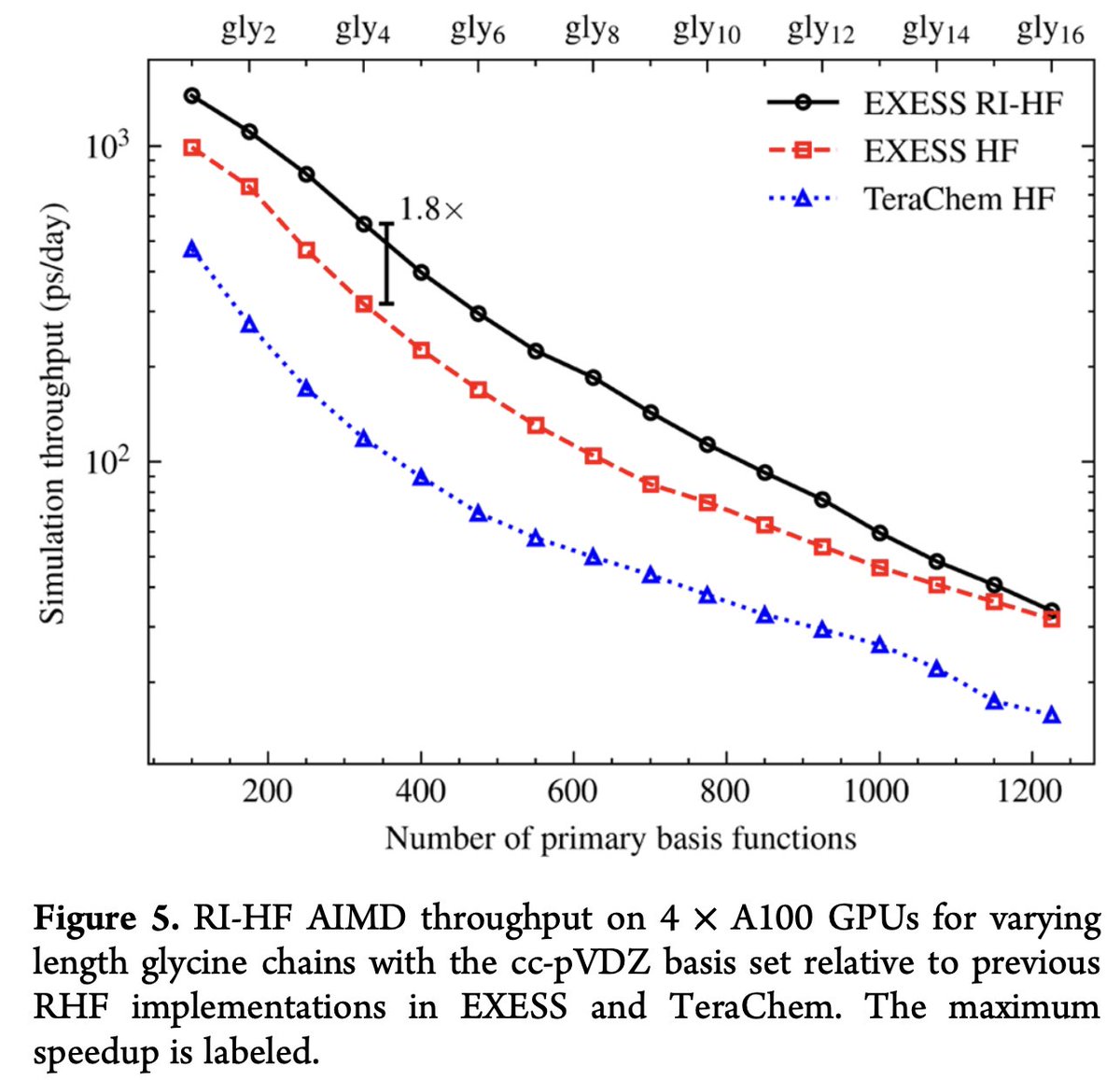
Sharing our latest paper, "Multi-GPU RI-HF Energies and Analytic Gradients─Toward High-Throughput Ab Initio Molecular Dynamics", now published in JCTC!
We introduce an optimized multi-GPU algorithm for high-throughput RI-Hartree-Fock energy, analytical gradients, and NVE AIMD calculations on small and medium molecules in EXESS. Our comparisons show that this is the fastest RI-HF implementation available. Data is provided up to quadrupole zeta basis sets.
https://t.co/ESVypPcg0l
#compchem #quantumphysics #gpu #HPC #AIMD
Thanks @ProfvLilienfeld. The current scoring function was designed to obtain a strong correlation with error in MBE expansions while jointly allowing to generate fragments with sizes normally distributed around a “desired” fragment size. The desired average fragment size is a user input. Thus, one other way to see it, it that for a given input average fragment size it generate fragment that allows to minimize MBE error. This also means that loosely the number of fragments is fixed by the user. Indeed, this is quite a good starting point for an ML approach for 1) generating directly fragments bypassing the genetic optimization (train a model to minimize the scoring function), 2) augment it to directly predict the error of MBE as you suggested. 2) requires extra thought and fine tuning to get quantitative accuracy, eg if one is targeting energies and gradients. However could be a nice starting point for even more interesting properties such as ligand-protein interactions …
Our latest paper on Quick Fragmentation via Automated Genetic Search (QFRAGS) is now on J. Cheminf.! 🎉 QFRAGS fully automates molecular fragmentation for biomolecules, minimizing energy errors with evolutionary optimization. Full paper: https://t.co/NKgUKjS43q #compchem#AI #cheminformatics