Husband, Dad, Professor/Director. Chemical Biology, Drug Discovery, TPD, PROTACs, Molecular Glues, ADCs, Proteomics, & AI/ML. We do 💻🧪🧬 🐁 Views are my own.
The @JinWangLab, H Lin et al developed a new analytical tool to optimize the potency and selectivity of drugs. #drugDesign@NatureComms@GENbio https://t.co/wrFZCeZ7Ph
@Eddie_Cliff@JAMA_current@amarkelkar@PORTAL_Research@JAMAOnc@JAMANetwork@suhas_gondi@bnrome Thanks for sharing the viewpoints, which are very much self-conflicting. It said MFN stifles innovation due to reduction of Pharma profits. On the other hand, it suggests to negotiate drug prices 1 year after approval, which will totally destroy Pharma and Biotechs profits. 🤥🙄
@AllThingsApx At this moment, docking is still way more useful than co-folding in identifying hits. In certain cases, such as chemoproteomics screen, co-folding could be useful. See our evaluation https://t.co/3Oxh0vzOQI
@BiologyAIDaily gave a much better summary than I did. 😂 Here is our manuscript evaluating AF3. Long story short, AF3 is a great start. But we still have much work to do to make it useful for drug discovery.
AlphaFold3 in Drug Discovery: A Comprehensive Assessment of Capabilities, Limitations, and Applications
1. This paper presents the first large-scale benchmarking of AlphaFold3 (AF3) for real-world drug discovery tasks, revealing that AF3 excels at predicting static protein-ligand interactions but struggles with dynamic conformational changes, GPCR state prediction, and affinity ranking.
2. In static binary complexes, AF3 achieves high structural accuracy and outperforms traditional docking in side-chain orientation recovery, making it ideal for generating initial complex models when minimal conformational change is expected.
3. AF3 demonstrates clear limitations in modeling induced-fit scenarios: performance significantly drops for dynamic complexes (RMSD > 5Å), and predictions decline on structures released after its training cutoff, suggesting overreliance on memorization.
4. AF3 consistently predicts active GPCR conformations, even when modeling antagonist-bound states, revealing a systematic conformational bias that persists despite extensive sampling.
5. The model fails to reliably generate ternary complex structures such as those found in PROTAC or molecular glue systems, showing poor agreement with experimental data and low protein–protein interaction recovery.
6. AF3's internal ranking metrics (score and PAE) show no meaningful correlation with experimental binding affinity across a panel of sEH inhibitors, confirming it cannot be used for lead prioritization or virtual screening ranking.
7. Despite these limitations, AF3 performs well in chemoproteomics applications, accurately identifying binding pockets in 82% of known targets and generating high-confidence structures that align closely with experimental data.
8. The study introduces a dual-filtration strategy (AF3 score > 0.7 + GNINA docking < -5 kcal/mol) to select high-quality protein–ligand models from AF3 outputs, demonstrating utility in fragment-to-lead design.
9. AF3 captures resistance-driving steric clashes in BTK mutants, with ranking scores reflecting binding disruptions seen in experimental KD values—though it lacks sensitivity to subtler electronic effects.
10. However, AF3 fails to predict kinase selectivity across the human kinome, with ROC-AUCs near random for three benchmark inhibitors, indicating its inability to resolve subtle binding preferences without steric conflict.
11. The paper concludes that AF3 functions best as a “true-hit binary interaction modeler,” useful for generating structures of validated binders but not yet suitable for affinity ranking, ternary prediction, or broad selectivity profiling.
12. Future improvements should integrate enhanced conformational sampling, physics-based scoring (e.g., FEP+), and training on more diverse structural states. Hybrid pipelines combining AF3 with molecular dynamics and docking are proposed as the way forward.
💻Code: https://t.co/EXPt6iZUKw
📜Paper: https://t.co/SpTfolUpCw
#AlphaFold3 #DrugDiscovery #StructuralBiology #GPCR #TernaryComplexes #ProteinLigand #BindingAffinity #Bioinformatics #PROTAC #Chemoproteomics #AI4Science
Congratulations to Xiangyu Deng and Yang Gao at @RiceUniversity for their outstanding work unraveling the complexities of RNA editing! Biochemical profiling and structural basis of ADAR1-mediated RNA editing: Molecular Cell https://t.co/vmR5vOJnkH
Interestingly, using the biochemical assays developed in this publication, we tested ZYS-1 (which I noted last week is not a genuine ADAR1 inhibitor) and confirmed it fails to inhibit ADAR1 even at concentrations as high as 1 mM.
Excited to share our new preprint on @biorxivpreprint , where we challenge the claim that ZYS-1 is a potent ADAR1 inhibitor, as reported by Wang et al. in @NatureCancer. Re: Concerns Regarding the Validation of ZYS-1 as a Bona Fide ADAR1 Inhibitor https://t.co/4ywUIoiroH
Our results revealed that ZYS-1 does not inhibit ADAR1 activity at concentrations up to 1 mM, challenging previous conclusions based solely on binding data.
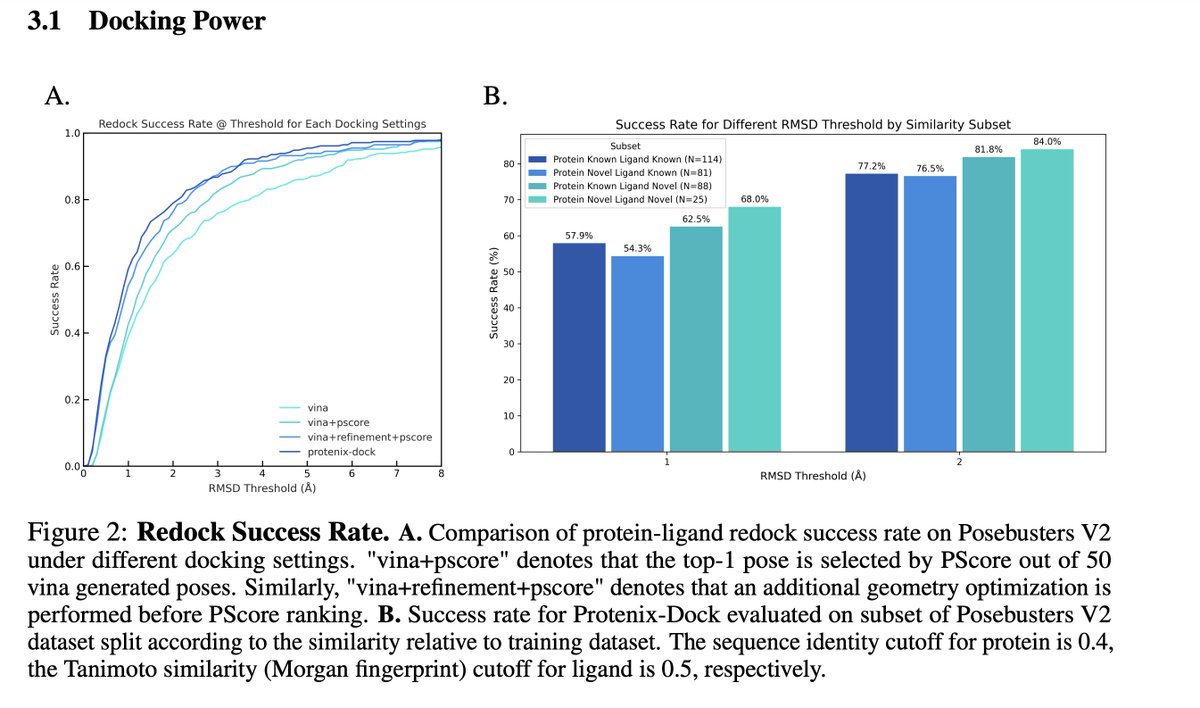
We just releasd our new docking tool: ProteniX-Dock ! Welcome everyone to try out~
GNU GPL3 so allows commercial use, and has a nice Python interface and CLI😁
Let's dive into it! 🧵
The NIH funds 4,400 active projects totaling $2.5 billion in Texas, much of it in Houston.
These cuts violate the law, hurt people, and undermine the very system of scientific research & groundbreaking advancements that we are so proud happen in #TX07.
https://t.co/WnhQWak1fu
Today, we at OpenAI launched Deep Researcher and I wanted to share a deeply personal story about how amazing this tool is and how it will change the world. Trigger warning, related to cancer....1/9