Our machine learning driven quantification algorithm for proteomics now published in Nature Biotechnology. Optimal protein quantification and, for the first time, error estimates for individual quantities. Paper link in comments below.
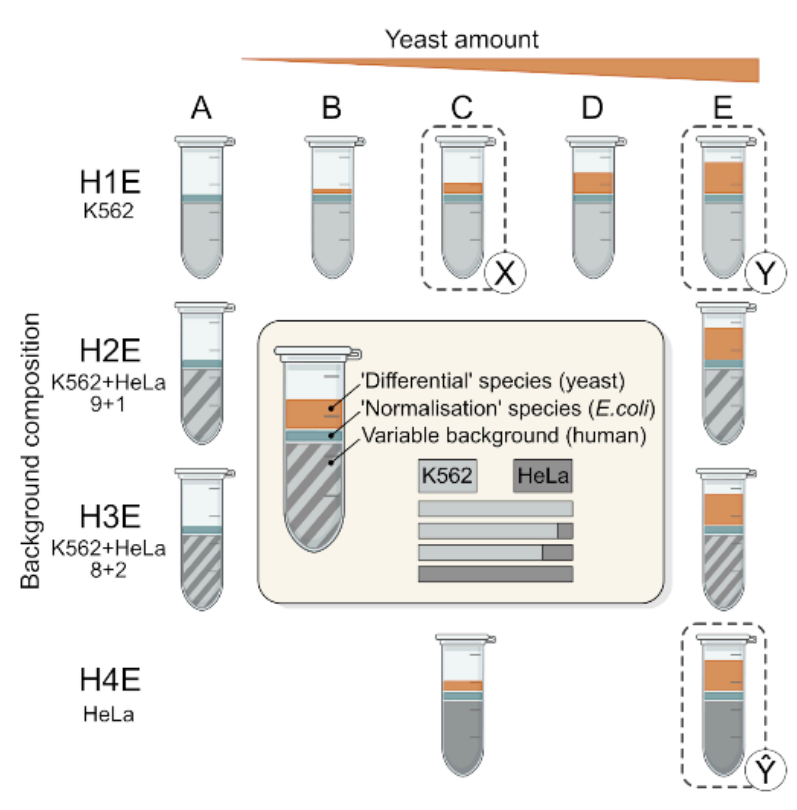
We acquired a large-scale mixed-species benchmark, with variable background, to comprehensively assess quantitative accuracy of proteomics.
Key features:
- 192 runs, 0.75ng - 15ng of variable human cell line backgroud.
- Can dissect the impact of both random and systematic errors.
- Can test how quantitation algorithms scale with experiment size and sample heterogeneity.
Our insights based on the data: https://t.co/53lZfcjitI.
PRIDE repo will be made public in the next days.
Solving the computational challenge of phosphoproteomics with 𝐏𝐡𝐨-𝐓𝐢𝐩: dephosphorylation on-tip identifies the sequences of phosphorylated peptides.
This serves as a basis for predicted spectral libraries, reducing the search space 10x-20x. https://t.co/1Si8QojpqR
We have now released 𝐃𝐈𝐀-𝐍𝐍 𝟐.𝟎!
DIA-NN 2.0 Academia: https://t.co/hxgTs5kYau.
We also now offer DIA-NN Enterprise for Industry, please contact Aptila Biotech https://t.co/QFYuxu7QCl.
Excited to announce DIA-NN version 1.9, our software suite for proteomics data processing!
A range of cool new features and general performance improvements. We consider it the most significant update in the history of DIA-NN.
https://t.co/RKzniysMLg
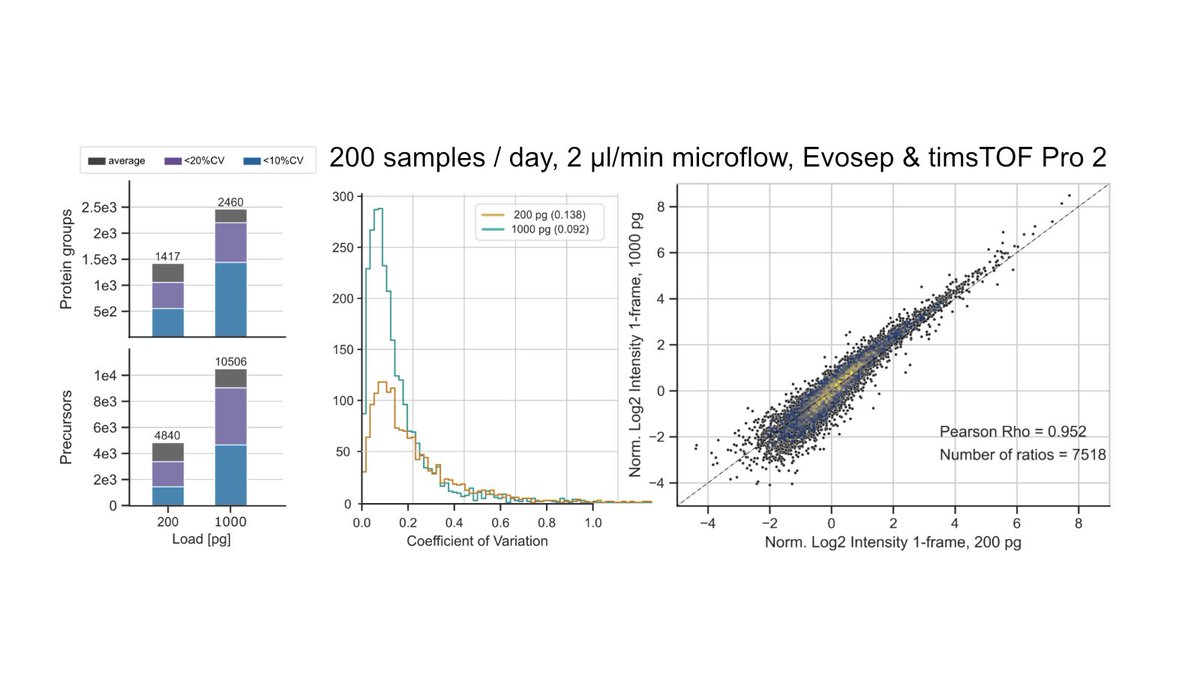
Want reliable quantities? Brilliant students in my lab @ZiskaKistner@JustusGrossmann figured out how. We present QuantUMS, an ML-based algorithm for step-change better quantitation in proteomics and statistical confidence in individual quantities https://t.co/9AhXScJfhl
QuantUMS is integrated in DIA-NN (beta version referenced in preprint), but we also plan to release it as an open-source tool, for use on various kinds of data, not just DIA.
We further anticipate significant gains to be achieved by integrating the accuracy metric reported by QuantUMS with existing packages for statistical analysis of proteomics data.
Some benchmarks are in the twitter thread below.
We present Slice-PASEF on timsTOFs from @BrukerMassSpec. Slice-PASEF can fragment all ions entering the mass spec and we show that it's by far the most sensitive discovery proteomics technology to date. Ideal for single-cell proteomics. https://t.co/a3dU3w7z4K
https://t.co/OmFQsSOaan Proudly presenting our preprint about OxoScan-MS, facilitating large scale (plasma) glycoprotemics, at high sensitivity and at low costs per samples #ASMS2022#TeamMassSpec
Massive collaboration with @OpenMSTeam and @DemichevLab to allow Quantitative MS-based data analysis distributed and in the cloud. quantms allows analyzing DDA-LFQ, DIA-LFQ, DDA-ISO (TMT) proteomics data.
https://t.co/pz6hhB1Kd8 Zeno SWATH, is our new variant of SWATH-MS, in which a linear ion trap is used to boost sensitivity in a DIA-MS experiment. We can now do #proteomics with fast high-flow chromatography only needing nanograms of sample. Great collaboration with @SCIEXOmics
What if the mass spec detector suddenly became 10x more sensitive? Proteomics of sub-microgram sample amounts now possible even on a high-flow LC @ 800 μl / min. Speed & sensitivity are now combined: Zeno SWATH @SCIEXnews, DIA-NN. https://t.co/FUb139cinF.