Researcher in CBIO team at @mines_paris and @institut_curie. Passionate about protein and RNA structure representation learning with geometric deep learning !
Learning Dynamic Protein Representations at Scale with Distograms
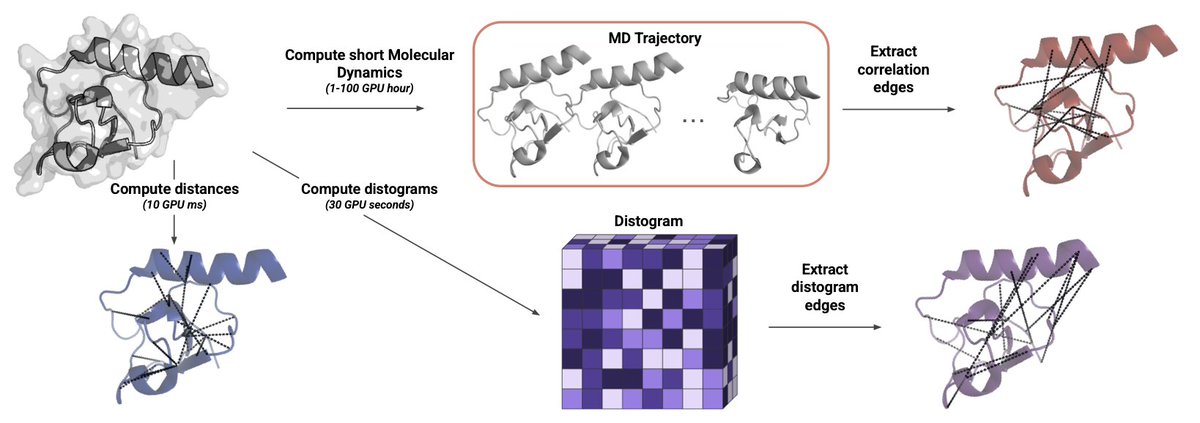
1. The authors bypass expensive MD by mining AlphaFold2/Boltz2 distograms—probability maps of residue–residue distances—to inject true conformational heterogeneity into graph neural networks without generating a single extra structure.
2. They turn each predicted distance distribution into a new edge set (Edisto) plus 64-bin edge features; a relational GNN then learns separate message-passing rules for static 3-D contacts, MD-derived motion correlations, and these cheap distogram links.
3. Across 24 architecture/task combinations distogram edges rank first 16× and second 8×; on ligand-binding-site prediction they raise F1 by up to 9.3 points versus MD-correlation graphs while needing orders-of-magnitude less compute.
4. The same trick boosts RNA tasks (chemical-modification and binding-site prediction) even though structure predictors are less accurate for RNA, hinting that uncertainty-encoded distances generalise beyond proteins.
5. When plugged into ThermoMPNN for ��∆G prediction, distogram edge features lift R² from 0.518 → 0.577 and Spearman ρ from 0.726 → 0.749 on the mega-scale stability dataset, again without any MD.
6. Overlap analysis shows distogram edges capture long-range “fuzziness” around loops whereas MD correlations highlight coordinated helix-sheet motions; the two signals are complementary but distograms are free once the structure predictor has run.
7. Limitations: quality still hinges on MSA richness, compute grows for huge complexes, and pairwise marginals can’t disentangle higher-order cooperativities—yet the route to large-scale dynamic modelling without MD is now open.
💻Code: https://t.co/gxleLN6LIj
📜Paper:https://t.co/4zuxGAydF8
#proteindynamics #AlphaFold #graphNN #drugdiscovery #structuralbiology #bioinformatics
@BiologyAIDaily We also propose a swapping-based experiment to assess the target-specifity of DTI methods. We applied this test to existing papers. To our surprise, some methods are quite specific ! 🙌
@BiologyAIDaily Not mentioned but important imo, we provide an assesment of the performance of Boltz2 for RNA-small molecule affinity prediction. SPOILER: it does not work, unfortunately.
It was an honor to contribute a small part to this expansive and highly insightful look at the current state of ML-assisted RNA drug design led by @MalletVincent and Wissam Karroucha.
Most exciting is a new benchmark on virtual screening specificity (including Boltz2).
https://t.co/jH2MjpoKvr
Leveraging Protein Representations to Explore Uncharted Fold Spaces with Generative Models
1. This novel study introduces DiffTopo, a novel coarse-grained protein structure representation method that uses diffusion models to efficiently explore uncharted areas of the protein fold space. By focusing on low-resolution topological sampling, it significantly enhances the discovery of novel protein folds beyond those found in nature.
2. The researchers combined DiffTopo with RFdiffusion, a backbone-level protein generative model, to rapidly generate novel protein folds. This integration allows for efficient exploration of the designable topology space, leading to the creation of 30 different novel topologies that were experimentally characterized, demonstrating the method's practical applicability.
3. An innovative aspect of this work is the MirrorTopo pipeline, which generates mirrored topologies of native proteins. These mirrored structures, while physically realistic, are absent from natural repertoires. The study successfully characterized 6 different novel mirror topologies, highlighting the potential for discovering entirely new protein architectures.
4. The study's experimental validation is particularly noteworthy. Out of 40 designed dark folds and 22 mirror folds, five crystal structures closely matched the computational models, confirming the high structural accuracy and designability of the generated proteins. This suggests that the method can access folds beyond the capabilities of current generative approaches.
5. The framework relies on a coarse-grained topology description that captures the spatial arrangements of α-helices and β-strands. This reduced-dimensionality approach smooths the sampling landscape, allowing for more diverse and novel fold discoveries. The separation between fold discovery (DiffTopo) and atomic-level sampling (RFdiffusion) overcomes limitations of existing methods that often recapitulate known topologies.
6. The study also explores the structural precision of the generated folds. For instance, the dark fold N30 features a particularly entangled topology with one helix threading through a closed cavity formed by two others. The successful crystallization and structural analysis of such complex designs highlight the potential for encoding nontrivial folding pathways.
7. The researchers further demonstrate the potential for functional design through motif scaffolding. By conditioning on existing secondary structure motifs, DiffTopo can generate diverse scaffolds that preserve motif geometry, expanding the possibilities for designing proteins with tailored interactions.
📜Paper: https://t.co/4TymOCYsrv
#ProteinDesign #GenerativeModels #ComputationalBiology #NovelFolds #MirrorTopologies
Our Ambient Protein Diffusion code is now available.
We trained a structure generation model on AlphaFoldDB that produces designable long proteins.
AlphaFoldDB contains proteins of varying quality. Our approach explicitly accounts for this during diffusion training.
A Comprehensive Benchmark for RNA 3D Structure-Function Modeling
1. This study introduces a comprehensive benchmarking suite for RNA structure-function modeling, addressing a significant gap in the field by providing datasets for various RNA-related tasks.
2. Seven RNA structure-function prediction tasks are proposed, covering areas like RNA function tagging (RNA-GO), molecular design (RNA-IF), and small molecule binding predictions (RNA-SITE, RNA-LIGAND), with novel dataset splits and evaluation methods.
3. The toolkit is built on the RNAgLib library, making it modular, reproducible, and easily accessible, promoting community contributions and easy customization for RNA-related deep learning tasks.
4. The authors present a set of datasets and tasks with clear data preprocessing strategies, ensuring high-quality and non-redundant RNA structures, which facilitate the comparison of different models for RNA 3D structure-function prediction.
5. Tasks like RNA-GO aim to predict RNA functions using Gene Ontology terms, while RNA-IF focuses on inverse folding, predicting sequences for given RNA structures. These tasks allow for the exploration of RNA's complex roles in cellular processes.
6. The benchmarking suite also includes tasks like RNA-CM for detecting chemically modified RNA residues, and RNA-PROT for predicting RNA-protein interactions, addressing diverse aspects of RNA structural biology.
7. For RNA drug design, the toolkit includes RNA-SITE for binding site detection and RNA-VS for virtual screening, aiming to accelerate drug discovery for RNA targets.
8. The authors demonstrate the utility of their benchmark by applying a simple graph neural network model on all tasks, providing initial baseline results that can serve as a reference for future model developments.
9. This benchmark is designed to be easily extended and adapted, with the potential for further integration of novel RNA tasks and representation learning techniques, driving advances in RNA structural biology and drug discovery.
10. By offering a standardized, reproducible framework, this work promises to significantly enhance the development and evaluation of deep learning models for RNA 3D structure-function prediction.
💻Code: https://t.co/oRVPS2t7My
📜Paper: https://t.co/MJCtAnMNpQ
#RNA #DeepLearning #Bioinformatics #MachineLearning #DrugDiscovery #RNA3DStructure #Benchmarking #StructuralBiology
Our latest efforts in AI-driven RNA drug discovery (RNAmigos2) have been published in Nature Communications.
Blessed to have worked with such a talented team: @MalletVincent, J. Waldispühl, JG Patiño, et al.
Paper: https://t.co/kv5Gqltqtj
GitHub: https://t.co/Yl5INi3bAf
Powered by: @rnaglib
Takeaways:
1. We achieve 10,000x speedup over docking at similar accuracies + boost ligand diversity over docking alone.
2. New benchmark tests generalization to new structures.
3. Successful zero-shot active enrichment on in-vitro assay.
4. Multi-modal data, self-supervision, and synthetic data are key.
5. Docking and AI models work well side-by-side if you have the budget.
@Oxer22 In addition, we show that instead of using these representations sequentially; sharing them across all layers improved results and stability, a possible future direction for S3F ?
@Oxer22 Great paper Zuobai ! Our paper AtomSurf https://t.co/N5MXCjKeXv also investigated the joint use of surface, graph and sequence, with a focus on protein interactions instead of protein fitness. It also reports better performance; maybe this combination is truly interesting ? 🤔
RNAmigos2: Fast and accurate structure-based RNA virtual screening with semi-supervised graph learning and large-scale docking data
1. RNAmigos2 changes RNA-targeted drug discovery with a machine-learning pipeline that outpaces traditional docking by running over 10,000 times faster while maintaining superior accuracy.
2. The model utilizes a novel 2.5D graph representation of RNA structures, capturing intricate base pair interactions to enhance binding site prediction and ligand screening.
3. By combining deep learning with docking data augmentation, RNAmigos2 achieves top 2.8% ranking for active compounds across structurally diverse test sets, proving its generalization capabilities.
4. The framework significantly improves virtual screening efficiency, screening up to 15.7 million compounds in a single day compared to 1,400 with traditional methods under similar compute budgets.
5. RNAmigos2 demonstrates experimental success by identifying RNA riboswitch ligands from a 20,000-compound assay with enrichment factors up to 5.09, marking a breakthrough in structure-based RNA drug discovery.
6. The model integrates seamlessly with docking techniques, forming RNAmigos++, which enhances enrichment scores while reducing computational costs, paving the way for hybrid approaches in RNA therapeutics.
@carlosgoliver@MalletVincent
💻Code: https://t.co/TkNdks3wDB
📜Paper: https://t.co/RCHMslJk1i
#RNAtherapeutics #DrugDiscovery #Bioinformatics #DeepLearning #VirtualScreening
AtomSurf: Surface Representation for Learning on Protein Structures
1/ AtomSurf introduces a novel surface representation approach for protein structure learning, significantly advancing previous methodologies by integrating surface and graph-based encoders for enhanced performance in protein tasks.
2/ The key innovation is the simultaneous learning of surface and graph-based features, which allows for a comprehensive understanding of protein structures. This hybrid approach ensures a more accurate prediction in tasks like binding site identification and protein interaction prediction.
3/ AtomSurf adapts the powerful DiffusionNet architecture for protein learning, optimizing it for efficiency and scale, resulting in competitive training and inference times compared to existing methods.
4/ This method achieves state-of-the-art performance across all tasks in the Atom3D benchmark, particularly excelling in protein-protein interaction prediction, mutation stability prediction, and protein structure ranking tasks.
5/ By allowing node-wise feature sharing between graph and surface representations, AtomSurf creates a synergy that outperforms traditional methods. It captures both the intricate surface geometries and the internal atomic interactions of proteins.
6/ The paper also showcases improvements in computational efficiency through the use of coarsened meshes and enhanced architecture design, making AtomSurf a highly effective tool in terms of both speed and memory usage.
7/ This approach has significant implications for structural bioinformatics and drug design, providing a more holistic way to model protein interactions, which could be critical in fields like antibody design and ligand-binding pocket classification.
💻Code: https://t.co/gTtq7ncHUQ
📜Paper: https://t.co/q8erAuyTLc