PhD Student | Computational biology | Structural bioinformatics | Machine Learning @ the #LinderstrømLang Centre for Protein Science @uni_copenhagen 🇩🇰
Are you analysing hydrophobicity in disordered proteins and still using something like the Kyte & Doolittle scale?
Maybe instead consider the "stickiness-scale" that Fan & Giulio derived using SAXS data for >100 IDPs
https://t.co/hN3KtuzsWh
A near-complete map of human cytosolic degrons and their relevance for disease
We measured degron potency of 212,658 30-residue tiles from 5,672 human proteins and trained a model to predict degrons from seq
Led by V Voutsinos in Hartmann-Petersen lab
https://t.co/KUBXAJg1Yc
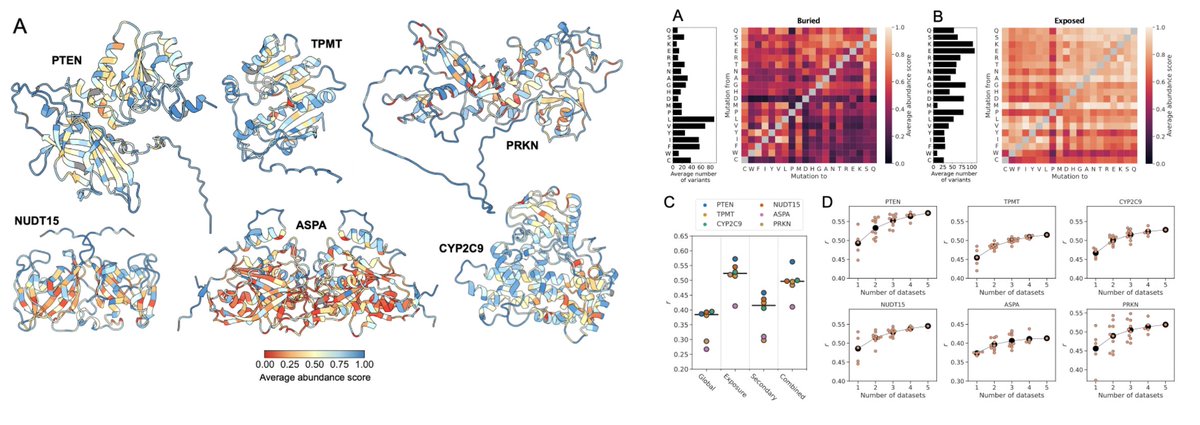
New preprint w @TKSchulze who analysed cellular abundance (VAMP-seq) data for ~32,000 variants of six proteins
We find that much of the variation can be explained and predicted by a burial-dependent substitution matrix
Lots more goodies in the paper
https://t.co/axsUhazLZ8
For a working version of the model and instructions on downloading our human proteome predictions, visit our GitHub repository: https://t.co/2c70kTUTgv
If you haven't seen it yet, check out our May 22 preprint! 🎉 Together with @CagiadaMatteo and @LindorffLarsen, we developed FunC-ESMs, a predictor separating loss of function variants caused by decreased stability from those affecting function by other means.
New preprint by @CagiadaMatteo and @NicolasJonsson who used ESM models to predict and analyse variant effects across the human proteome and separated loss of function variants into those caused by decreased stability from those affecting "functional sites"
https://t.co/RrYFQtBDo9
Finally, we analyzed clinically annotated variants to determine if their pathogenicity was due to loss of function in the protein, and whether this loss of function was driven by decreased stability.
Very happy to share work led by @SoBuelow on prediction of phase separation propensities of disordered proteins from sequence
We combined active learning and coarse-grained simulations to develop a machine learning model for quantitative predictions of IDR phase separation
Matteo will present the results from a new manuscript (by @CagiadaMatteo and @NicolasJonsson) at #VariantEffect24 🧬 🖥️ . Preprint sitting in the biorxiv queue and will hopefully be released by tomorrow 😊
New preprint with @CagiadaMatteo & @sokrypton in which we present a benchmark and predictions of absolute protein stability (ΔG not ΔΔG) using using likelihoods from a generative model, and also benchmark it for conformational free energies against NMR
https://t.co/B7Pp5FJQiF
Happy to share our mutational atlas for Parkin proteostasis
We measured the abundance of ~9000 missense variants of Parkin, and show how many unstable variants are protasomal targets, incl. some that form neo-degrons in an exposed hydrophobic loop
https://t.co/D867T2I5Vp
Happy to share the publication of our paper in @Nature
Conformational ensembles of the human intrinsically disordered proteome
Work led by @GiulioTesei and @AnnaIdaTrolle. I'll post more later, but for now here is a link:
https://t.co/b3aZ6BbGpB
and a short movie about the work
Do you enjoy interdisciplinary science and are looking for a PhD position? Then consider the positions in our #DynaPLIX ERC Synergy project to use MD simulation 🖥️ with NMR 🧲 & time-resolved crystallography ⏱️⚡️ to study how ligands 💊 bind to proteins
https://t.co/Fx4lsfBrJ4
Come join us at the Department of Biology, University of Copenhagen 🇩🇰🌞🌦️🧬🍝🧶🧲🧫💻
We have wonderful colleagues 😊 and students
We are looking to hire a new group leader, and can fill the position at either of these levels: TT/Assoc/Full professor
📣 https://t.co/sUonJ3eeyo
New preprint led by Lene Clausen and Vasileios Voutsinos in @rasmushartmannp lab in which we determined and analysed effects of essentially all missense variants in Parkin (PARK2) on protein abundance using VAMP-seq incl. effects of an exposed degron 🧬🧠
https://t.co/sPNF4mR2hy
Do you like ∆∆G values as much as we do? In that case, you might like the >10^8 stability changes that we calculated for essentially all possible single amino acid changes in the human proteome. All described in @blaabjerg_lasse's paper on RaSP https://t.co/JLzNHnOPf2
Now out:
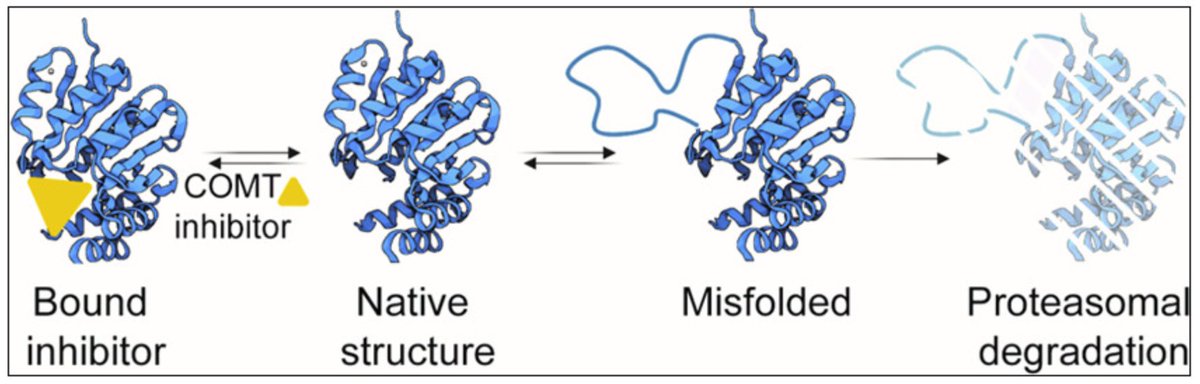
Rare Catechol-O-methyltransferase Missense Variants Are Structurally Unstable Proteasome Targets
with Fia Larsen, @CagiadaMatteo, Jonas Dideriksen, @_amelie_rocks and @rasmushartmannp
https://t.co/phPTkXUAUy
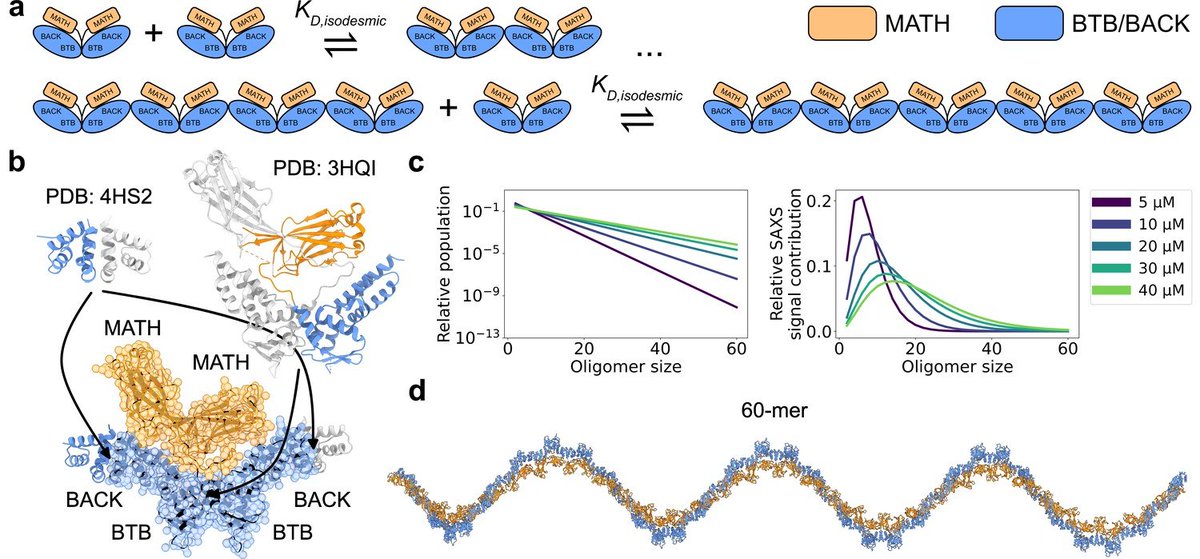
How do you analyse SAXS data of a system when it shows both conformational and compositional heterogeneity?
For the answer, see work led by @EmilThomasen out in @eLife, where we determine conformational ensembles of the self-associating protein SPOP. 1/n
https://t.co/Hd8tvYc6ev
Great to learn of many researchers and companies using the data from our Mega-scale folding stability paper! If you are using these data it would be fantastic if you could register here https://t.co/UVu0CL57CT so we can show funders why this is important!