Professor of Cell Biology at Universitat Politècnica de València, with a particular passion for molecular medicine and a strong interest in natural philosophy.
Molecular Medicine is not just a branch of science — it is the very language of life.
It explains how cells communicate, how genes express identity, and how molecules decide between health and disease.
Within its scope lies the most intimate narrative of our existence: the silent choreography of enzymes, receptors, and signals that sustain us every second.
By following this account, you will explore the hidden logic that keeps us alive — and discover what happens when those delicate mechanisms fail, giving rise to illness.
Here, we decode the molecular origins of disease and the rationale behind the action of drugs, tracing every therapeutic effect back to its biochemical root.
Understanding Molecular Medicine is to see medicine itself under a microscope — where every cure begins as a molecular idea.
Here is the definitive guide to understanding something you have heard many times, but perhaps never fully understood: why statins can, in rare cases, damage muscle.
🌟 Statins rarely produce true rhabdomyolysis, but when they do, the core problem is not simply “muscle toxicity”.
When statins inhibit HMG-CoA reductase, they touch a metabolic node that connects lipid metabolism, mitochondrial respiration, intracellular trafficking and calcium homeostasis.
🚩 This pathway does not only produce cholesterol.
It also produces several molecules essential for muscle-cell function, including ubiquinone/coenzyme Q10, isoprenoids, and intermediates required for protein prenylation.
This is why the adverse effect is molecularly broader than “low cholesterol in muscle”.
✳️ At the mitochondrial level, reduced mevalonate-derived products may impair electron transport, especially through effects on oxidative phosphorylation.
The myocyte then produces less ATP and more oxidative stress.
Skeletal muscle is particularly vulnerable because calcium handling, membrane repair, contraction–relaxation cycling, and proteostasis are all ATP-demanding processes.
When ATP falls, the sarcoplasmic reticulum and plasma membrane cannot maintain ionic gradients efficiently.
✳️ A second mechanism involves isoprenoid depletion.
Small GTPases such as Rho, Rac, Rab and Ras require prenylation to anchor correctly to membranes and regulate vesicular trafficking, cytoskeletal organisation, autophagy, and survival signalling.
Statins can therefore disturb membrane dynamics and intracellular logistics, not just lipid synthesis.
✳️ Then comes calcium.
Mitochondrial stress and defective sarcoplasmic reticulum regulation favour cytosolic Ca²⁺ accumulation.
Calcium activates proteases such as calpains, phospholipases, and mitochondrial permeability transition.
The fibre begins to digest itself from within. If the damage remains limited, the patient experiences myalgia or mild CK elevation.
✳️ If the process becomes extensive, sarcolemmal integrity fails and the myocyte releases creatine kinase, myoglobin, potassium, phosphate and intracellular enzymes into the circulation. That is rhabdomyolysis.
✳️ Risk increases when muscle exposure to statins rises: high doses, lipophilic statins, renal or hepatic impairment, hypothyroidism, advanced age, intense exercise, and drug interactions, especially with CYP3A4 inhibitors or gemfibrozil.
✳️ Genetic variants also matter, particularly SLCO1B1, which encodes the hepatic uptake transporter OATP1B1; reduced hepatic uptake can increase circulating statin levels and muscle exposure.
➡️ So, mechanistically, statin-induced rhabdomyolysis is best understood as a convergence of:
mevalonate blockade → mitochondrial dysfunction + impaired prenylation + calcium dysregulation → proteolysis, membrane rupture and myocyte necrosis.
🗺️ ¿Cómo es el relieve de Andalucía? Un recorrido espectacular en relieve 3D por sus 8 provincias en orden alfabético. Abrimos hilo para descubrir la geografía andaluza detalle a detalle. ¡Búscate y comparte tu provincia! 👇
Recently, in @NEJM a phase 1 trial reported the first clinical data with VERVE-102, a therapy that changes the logic of PCSK9 inhibition.
VERVE-102 is not a conventional PCSK9 inhibitor. It is an in vivo adenine base-editing therapy designed to durably inactivate PCSK9 in hepatocytes after a single intravenous infusion. It contains an mRNA encoding an adenine base editor and a guide RNA targeting PCSK9, packaged in a GalNAc-lipid nanoparticle to favour liver uptake.
Why does this matter?
Because PCSK9 is one of the clearest examples of how human genetics can become pharmacology.
PCSK9 is produced mainly by the liver and controls the fate of the LDL receptor.
When PCSK9 binds LDL receptors, it promotes their degradation.
Fewer LDL receptors on hepatocytes means less LDL cholesterol removed from plasma.
The consequence is chronic exposure of the arterial wall to atherogenic lipoproteins.
But individuals with natural loss-of-function variants in PCSK9 show the opposite phenotype: lower LDL cholesterol and fewer atherosclerotic cardiovascular events.
VERVE-102 tries to recreate that protective biology therapeutically.
The mechanism is elegant.
After intravenous infusion, the GalNAc-lipid nanoparticle is taken up by hepatocytes, partly through the asialoglycoprotein receptor.
Inside the cell, the mRNA is translated into an adenine base editor, which pairs with the guide RNA and enters the nucleus. There, it targets a splice site near the beginning of PCSK9.
Unlike classical CRISPR nucleases, this system does not create a double-strand DNA break.
It performs a single chemical correction: adenine is converted into inosine, which the cell reads as guanine. The result is an A·T to G·C substitution that disrupts PCSK9 expression by introducing abnormal splicing and a premature stop codon.
Less PCSK9 means more LDL receptor recycling, more LDL clearance, and lower circulating LDL cholesterol.
In the trial, the highest dose reduced circulating PCSK9 by a mean of 88% and LDL cholesterol by 62%, with reductions appearing sustained during follow-up.
The conceptual shift is profound: this is not simply blocking a protein; it is attempting to convert a high-risk lipid phenotype into a genetically protected one.
@AnupamKhemariya insulin resistance arises when adipose storage, mitochondrial oxidation, inflammatory control and hormonal signalling become overwhelmed or dysregulated. It is less a single defect than a systemic failure of metabolic flexibility.
Recently, in @NEJM a phase 1 trial reported the first clinical data with VERVE-102, a therapy that changes the logic of PCSK9 inhibition.
VERVE-102 is not a conventional PCSK9 inhibitor. It is an in vivo adenine base-editing therapy designed to durably inactivate PCSK9 in hepatocytes after a single intravenous infusion. It contains an mRNA encoding an adenine base editor and a guide RNA targeting PCSK9, packaged in a GalNAc-lipid nanoparticle to favour liver uptake.
Why does this matter?
Because PCSK9 is one of the clearest examples of how human genetics can become pharmacology.
PCSK9 is produced mainly by the liver and controls the fate of the LDL receptor.
When PCSK9 binds LDL receptors, it promotes their degradation.
Fewer LDL receptors on hepatocytes means less LDL cholesterol removed from plasma.
The consequence is chronic exposure of the arterial wall to atherogenic lipoproteins.
But individuals with natural loss-of-function variants in PCSK9 show the opposite phenotype: lower LDL cholesterol and fewer atherosclerotic cardiovascular events.
VERVE-102 tries to recreate that protective biology therapeutically.
The mechanism is elegant.
After intravenous infusion, the GalNAc-lipid nanoparticle is taken up by hepatocytes, partly through the asialoglycoprotein receptor.
Inside the cell, the mRNA is translated into an adenine base editor, which pairs with the guide RNA and enters the nucleus. There, it targets a splice site near the beginning of PCSK9.
Unlike classical CRISPR nucleases, this system does not create a double-strand DNA break.
It performs a single chemical correction: adenine is converted into inosine, which the cell reads as guanine. The result is an A·T to G·C substitution that disrupts PCSK9 expression by introducing abnormal splicing and a premature stop codon.
Less PCSK9 means more LDL receptor recycling, more LDL clearance, and lower circulating LDL cholesterol.
In the trial, the highest dose reduced circulating PCSK9 by a mean of 88% and LDL cholesterol by 62%, with reductions appearing sustained during follow-up.
The conceptual shift is profound: this is not simply blocking a protein; it is attempting to convert a high-risk lipid phenotype into a genetically protected one.
CO2 Transport in Blood
Carbon dioxide undergoes physical dissolution and diffuses into adjacent blood capillaries. A small portion of CO2 in the blood remains dissolved, while the rest is chemically bound in form of HCO3 and carbamate residues of haemoglobin.
Ernst Haeckel made great contributions in the field of biology and his legacy as scientific illustrator is extraordinary. His master work "Art forms of Nature" influenced not only in science, but in the art, design and architecture of the early 20th century. Here, some sea anemones classified as Actiniae.
Insulin resistance is often reduced to “high glucose”. But that is only the surface of the problem.
Insulin is not simply a hormone that lowers blood sugar.
It is a systemic anabolic signal.
After a meal, insulin tells the body that nutrients are available and that energy can be stored, used, or redirected.
In skeletal muscle, insulin promotes glucose uptake through GLUT4 translocation.
This is one of its most familiar actions: glucose leaves the bloodstream and enters muscle fibres, where it can be oxidised or stored as glycogen.
In the liver, insulin suppresses glucose production.
It inhibits gluconeogenesis and glycogen breakdown, while favouring glycogen synthesis. In other words, after eating, the liver should stop behaving as if the body were fasting.
In adipose tissue, insulin inhibits lipolysis.
It tells fat cells not to release fatty acids into the circulation, because energy is already abundant. At the same time, it favours lipid storage.
But insulin also regulates protein metabolism, vascular tone, mitochondrial function, inflammation, and cellular growth pathways. It is not a “glucose hormone”; it is a metabolic coordinator.
Insulin resistance appears when tissues no longer respond adequately to that signal.
The pancreas compensates by secreting more insulin, often for years. This creates a paradox: insulin levels are high, but insulin action is incomplete.
The consequence is not uniform failure.
Some insulin pathways become resistant, while others remain active. Glucose uptake in muscle may fall, and hepatic glucose production may remain inappropriately high. Yet lipogenesis in the liver can continue, contributing to fatty liver and hypertriglyceridaemia.
Meanwhile, adipose tissue releases more fatty acids, feeding hepatic fat accumulation and interfering with insulin signalling in muscle and liver. Chronic low-grade inflammation, ectopic lipid deposition, mitochondrial stress, and adipokine imbalance amplify the loop.
This is why insulin resistance is not just a prelude to type 2 diabetes. It is a whole-body disorder of nutrient partitioning.
Insulin resistance means that the body hears abundance, but its tissues behave as if the message were distorted. The problem is not only excess sugar in blood, but a loss of metabolic synchrony.
CO2 Transport in Blood
Carbon dioxide undergoes physical dissolution and diffuses into adjacent blood capillaries. A small portion of CO2 in the blood remains dissolved, while the rest is chemically bound in form of HCO3 and carbamate residues of haemoglobin.
Tumour suppressor genes constitute a central defensive layer against malignant transformation, acting to restrain inappropriate cell proliferation and preserve genomic stability.
🌟Their inactivation is a defining event in carcinogenesis, not because it confers a novel function, but because it removes critical biological constraints that normally limit cellular behaviour.
🌟 Traditionally, tumour suppressors have been divided into two functional classes: gatekeepers and caretakers.
▶️ Gatekeeper genes directly regulate cell fate decisions such as proliferation, senescence, differentiation, and apoptosis.
▶️ Prototypical examples include TP53 and RB1. When functioning normally, these genes act as molecular brakes, integrating signals related to DNA damage, oncogene activation, or cellular stress, and halting cell cycle progression when conditions are unfavourable.
▶️ Loss-of-function mutations in gatekeepers lead to transformation by releasing these brakes, allowing cells to proliferate despite accumulated damage or inappropriate growth signals. In this sense, gatekeepers exert immediate control over tumour initiation at the level of individual cells.
▶️ Caretaker genes, in contrast, do not directly regulate proliferation. Instead, they are responsible for maintaining the integrity of the genome through DNA repair, replication fidelity, and chromosomal stability.
▶️ Genes involved in mismatch repair, homologous recombination, or nucleotide excision repair fall into this category.
▶️ Defects in caretakers generate a so-called “mutator phenotype”, characterised by an accelerated accumulation of mutations across the genome.
▶️ Importantly, caretaker gene loss does not directly drive proliferation; rather, it creates the permissive conditions for mutations in gatekeepers and oncogenes to arise. This explains why patients with inherited defects in DNA repair pathways, such as those with mismatch repair deficiency, exhibit a markedly increased cancer risk across multiple tissues.
▶️ Beyond this binary classification, a third functional concept has gained relevance: landscape or “landscaper” tumour suppressor genes.
▶️ These genes influence the tissue microenvironment rather than the intrinsic behaviour of epithelial cells. By altering stromal composition, extracellular matrix organisation, or inflammatory signalling, landscapers indirectly promote tumour development by creating a permissive niche for malignant progression.
▶️ Together, these categories highlight that tumour suppression is not a single mechanism but a coordinated, multi-layered system.
▶️ Cancer emerges not simply from increased proliferation, but from the progressive dismantling of safeguards that normally preserve genomic fidelity, tissue architecture, and controlled cellular behaviour.
Tumour suppressor genes constitute a central defensive layer against malignant transformation, acting to restrain inappropriate cell proliferation and preserve genomic stability.
🌟Their inactivation is a defining event in carcinogenesis, not because it confers a novel function, but because it removes critical biological constraints that normally limit cellular behaviour.
🌟 Traditionally, tumour suppressors have been divided into two functional classes: gatekeepers and caretakers.
▶️ Gatekeeper genes directly regulate cell fate decisions such as proliferation, senescence, differentiation, and apoptosis.
▶️ Prototypical examples include TP53 and RB1. When functioning normally, these genes act as molecular brakes, integrating signals related to DNA damage, oncogene activation, or cellular stress, and halting cell cycle progression when conditions are unfavourable.
▶️ Loss-of-function mutations in gatekeepers lead to transformation by releasing these brakes, allowing cells to proliferate despite accumulated damage or inappropriate growth signals. In this sense, gatekeepers exert immediate control over tumour initiation at the level of individual cells.
▶️ Caretaker genes, in contrast, do not directly regulate proliferation. Instead, they are responsible for maintaining the integrity of the genome through DNA repair, replication fidelity, and chromosomal stability.
▶️ Genes involved in mismatch repair, homologous recombination, or nucleotide excision repair fall into this category.
▶️ Defects in caretakers generate a so-called “mutator phenotype”, characterised by an accelerated accumulation of mutations across the genome.
▶️ Importantly, caretaker gene loss does not directly drive proliferation; rather, it creates the permissive conditions for mutations in gatekeepers and oncogenes to arise. This explains why patients with inherited defects in DNA repair pathways, such as those with mismatch repair deficiency, exhibit a markedly increased cancer risk across multiple tissues.
▶️ Beyond this binary classification, a third functional concept has gained relevance: landscape or “landscaper” tumour suppressor genes.
▶️ These genes influence the tissue microenvironment rather than the intrinsic behaviour of epithelial cells. By altering stromal composition, extracellular matrix organisation, or inflammatory signalling, landscapers indirectly promote tumour development by creating a permissive niche for malignant progression.
▶️ Together, these categories highlight that tumour suppression is not a single mechanism but a coordinated, multi-layered system.
▶️ Cancer emerges not simply from increased proliferation, but from the progressive dismantling of safeguards that normally preserve genomic fidelity, tissue architecture, and controlled cellular behaviour.
A standard serum B12 test can appear normal while the body is actually functionally deficient at the cellular level. Two deeper markers reveal the truth:
•Homocysteine
•MMA
Even with levels nearly 3× above the deficiency cutoff, functional deficiency is still possible
We owe Kary Mullis a great debt for his crucial invention of PCR. It remains one of the most important techniques in modern biology and medicine.
That said, Mullis was a cretin.
Receiving a Nobel Prize does not automatically make someone a cultivated or wise person. His Nobel recognized a brilliant but essentially technical achievement—the practical insight to cycle enzymatic amplification—rather than any deep or original reasoning about biology or disease.
In my opinion his statement is not right. When PCR is performed properly in a clean, non-contaminated environment, with specific primers, appropriate cycle numbers, and negative controls, trace DNA does not contaminate the reaction or produce false-positive results. The method is highly specific under correct laboratory conditions.
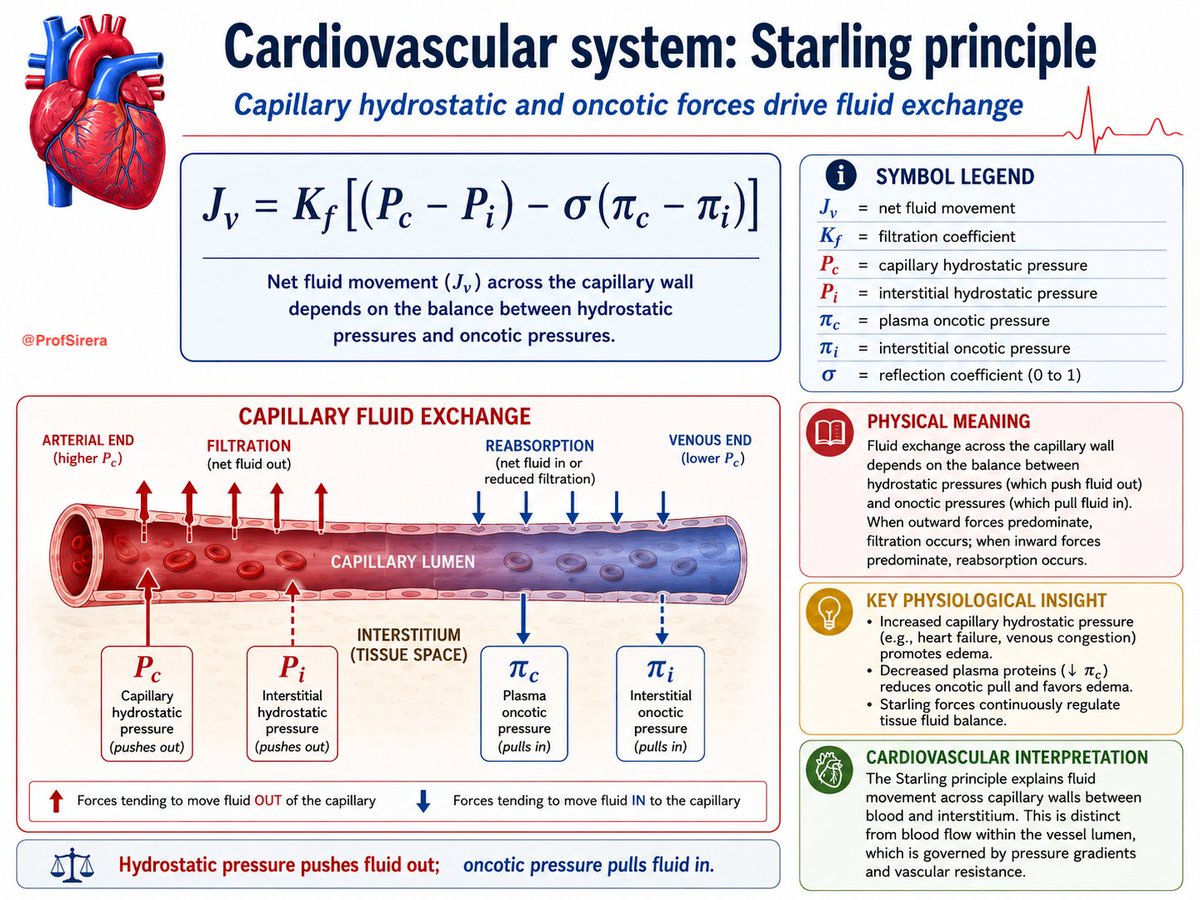
Starling’s principle explains capillary fluid exchange.
It is a balance between hydrostatic pressure, which pushes fluid out of capillaries, and oncotic pressure, mainly generated by plasma proteins, which pulls fluid back in.
Filtration predominates at the arterial end; reabsorption and lymphatic drainage prevent tissue fluid accumulation.
Inflammation disrupts this balance.
Dear @docakx, it does not seem that this post has attracted the attention of some of the other very popular topics on your account. What a pity—because biologically it is extraordinarily interesting.
Solar lentigines are not simple accumulations of pigment. They are localised territories of chronic photobiological remodelling.
The first misconception is to think that ultraviolet radiation merely “stimulates melanin”.
The reality is far more complex.
Chronic UV exposure continuously injures keratinocytes, damages mitochondrial and nuclear DNA, generates reactive oxygen species, oxidises membrane lipids, and activates inflammatory transcriptional programmes.
Over time, this alters the dialogue between keratinocytes, fibroblasts and melanocytes.
One of the central events is the persistent activation of paracrine melanogenic signalling.
UV-damaged keratinocytes increase secretion of α-MSH, endothelin-1 and stem cell factor, which chronically stimulate melanocytes, maintaining melanocyte activation even after the acute UV exposure has ended.
Interestingly, solar lentigines do not necessarily contain dramatically more melanocytes.
The key alteration is functional hyperactivity. These melanocytes produce more melanin, transfer melanosomes more efficiently, and operate within an epidermis whose architecture has already changed through photoageing.
The dermis participates as well.
Photoaged fibroblasts become senescent and begin secreting inflammatory mediators, growth factors and matrix-remodelling enzymes, continuously feeds abnormal instructions to the epidermis above it.
This explains why solar lentigines behave differently from freckles.
Freckles are largely genetically determined and fluctuate seasonally. Solar lentigines are stable “microfields” of cumulative photodamage. They represent decades of oxidative stress biologically recorded into the skin.
What appears clinically as a small brown macule is therefore not merely pigment.
It is a visible archive of UV-induced molecular ageing.
We owe Kary Mullis a great debt for his crucial invention of PCR. It remains one of the most important techniques in modern biology and medicine.
That said, Mullis was a cretin.
Receiving a Nobel Prize does not automatically make someone a cultivated or wise person. His Nobel recognized a brilliant but essentially technical achievement—the practical insight to cycle enzymatic amplification—rather than any deep or original reasoning about biology or disease.
In my opinion his statement is not right. When PCR is performed properly in a clean, non-contaminated environment, with specific primers, appropriate cycle numbers, and negative controls, trace DNA does not contaminate the reaction or produce false-positive results. The method is highly specific under correct laboratory conditions.