Don't Rush into Low-dose transdermal nicotine!
1. Watch our YouTube videos
2. Join the private FaceBook group for support
3. Go slow and low...
https://t.co/ou6ZTwPOEd
I think in some patients both ME/CFS and Long COVID may start from the same root problem: persistent antigen or viral persistence keeping the immune system chronically activated and, over time, triggering GPCR autoimmunity.
That said, nicotine is probably not directly fixing anti-muscarinic autoimmunity itself.
What seems more likely is that it helps indirectly in two ways. First, nicotinic receptors are involved higher up in autonomic signaling, at the level of the autonomic ganglia, before the final muscarinic signal reaches target organs. So if you boost nicotinic signaling, you may partially improve autonomic output even if the downstream muscarinic pathway is still dysfunctional.
Second, nicotine can also activate the α7 nicotinic anti-inflammatory pathway, which may dampen part of the neuroimmune amplification in some patients.
That also fits the antibody literature better, because in ME/CFS and Long COVID the most repeated findings are not really nicotinic receptor autoantibodies, but mainly β-adrenergic and muscarinic ones, especially β2, M3 and M4.
So my view is that low-dose nicotine may help a subgroup with a more cholinergic or autonomic phenotype, especially those with autonomic receptor dysfunction or related autoantibodies. But patients who do not have that kind of autonomic autoantibody-driven imbalance probably would not be expected to benefit much, because nicotine would not be acting on their main disease mechanism.
@Dfrizz007@HustleBitch_ We have smokers in our group who have benefited from nicotine patches and were able to stop smoking. They win on both. Nicotine through the patch is much healthier than smoking and has other benefits.
Researchers detected the following:
- Bartonella (a bacterium often spread by fleas or ticks) in 11 (22%)
- Babesia (a tick-borne parasite) in 10 individuals (20%)
- and both in 2 (4%)
Overall, nearly half (46%) showed evidence of at least one pathogen in their blood.
Read more: https://t.co/Ozo7usgjrd
Interested to know whether people have had improvements in PEM from mast cell MCAS medication.
H1/H2 blockers
Mast cell stabilizers
Xolair/omalizumab
Imatinib
Dupilumab
Etc
"Dopamine-based therapies could serve as a promising new direction for treatment"
@TheNicotineTest
IMO, low-dose nicotine can help, but dose needs to be flexible.
New study identifies dopamine's role in reversing Alzheimer's https://t.co/ueNyEFEWBM
🐀🐁🐀 Stage,
but on the right path!
Low-dose nicotine has a similar effect on the same receptors. It also increases CSF flow.
@TheNicotineTest
Scientists reverse Alzheimer’s in mice with breakthrough nanotechnology | ScienceDaily https://t.co/O4di6xWbOk
Prolactin and Nicotine.
Low-dose = minimal effect
high single doses = acute prolactin spikes
Chronic use = reduced prolactin
Since nicotine affects prolactin, it might be useful to look at them together.
New video!
Tom Bunker @BiologistBunk and I dig into the messy topic of research measures and trial designs.
We will all crack this code together!
https://t.co/DqBKhAlnqP
There’s often confusion about what subgroups are for in diseases. They’re not to say “this patient has it, this one doesn’t.” They’re to ask: among people who have it, why do symptoms differ, and what works for whom?
Cancer is the the most mature example. Breast cancer has 4 defined subgroups (Luminal A, Luminal B, HER2-enriched, Triple-Negative), each with its own treatment pathway.
These subgroups exist precisely because patients weren’t responding uniformly to treatment. Researchers needed to understand why nonresponders weren’t responding, and what they would respond to. Without that work, deaths attributed to breast cancer would be far higher today.
Subgrouping isn’t about classifying different diseases. It’s about identifying different trajectories and treatment responses within the same one. Every disease deserves it.
Scientists just found a way to reverse osteoporosis – not just slow it.
Researchers from the University of Leipzig and Shandong University have identified GPR133, a cell-surface receptor that functions as a master regulator of bone-building cells called osteoblasts. When GPR133 is switched on, osteoblasts ramp up activity and dramatically increase bone density.
In mouse experiments, animals engineered without the GPR133 gene developed fragile, porous bones similar to human osteoporosis. However, treating them with a synthetic activator called AP503—a small molecule discovered through computational screening—rapidly strengthened their skeletons. Remarkably, AP503 not only boosted bone mass in healthy mice but also reversed osteoporosis-like damage in older or diseased animals, with even greater effects when paired with physical exercise.
Because GPR133 influences bone density in humans and the underlying molecular pathways are highly conserved across mammals, the findings raise hope for a breakthrough osteoporosis therapy. Unlike existing drugs that merely slow bone loss (and often carry side effects or waning efficacy), a GPR133-activating compound could actively rebuild bone from within.
This approach could be particularly transformative for postmenopausal women, who face the highest risk of debilitating fractures, offering a safer, more effective way to restore skeletal strength.
["The mechanosensitive adhesion G protein-coupled receptor 133 (GPR133/ADGRD1) enhances bone formation." Signal Transduction and Targeted Therapy, 30 June 2025]
Neurodegeneration Is a Systems Failure — Not Just Neuron Loss
For decades, brain diseases like Alzheimer’s were framed as a linear story: neurons accumulate toxic proteins, then die. But converging evidence across high-impact studies suggests a different model — one that is far more integrated, and clinically actionable.
A recent study in Proceedings of the National Academy of Sciences (DOI: 10.1073/pnas.2516601123) demonstrates that during sleep, neural slow waves, vascular oscillations, and cerebrospinal fluid (CSF) flow are tightly coupled. This coordinated system drives glymphatic clearance — effectively a “wash cycle” for the brain. Disruption of this coupling may impair the removal of metabolic waste, including amyloid-β, linking sleep physiology directly to neurodegenerative risk.
At the cellular level, a landmark atlas published in Science (DOI: 10.1126/science.adf6812) reveals that vulnerability in the human brain is highly cell-type specific. Rather than uniform degeneration, distinct neuronal subpopulations exhibit selective susceptibility depending on their molecular identity, metabolic state, and anatomical context. This reframes neurodegeneration as a problem of selective circuit failure, not global decline.
Meanwhile, vascular integrity is emerging as a central determinant of disease progression. Work in Nature Medicine (DOI: 10.1038/s41591-023-02523-3) shows that aging is associated with breakdown of the blood–brain barrier (BBB), enabling peripheral immune cells and inflammatory mediators to infiltrate the central nervous system. This creates a feed-forward loop of neuroinflammation, exacerbating neuronal dysfunction.
Finally, synaptic loss — not neuron death — appears to be the earliest pathological event. Evidence from Nature Neuroscience (DOI: 10.1038/s41593-022-01186-2) indicates that activated microglia mediate excessive synaptic pruning through complement pathways. This process dismantles neural circuits before neurons themselves are lost, suggesting that functional decline begins much earlier than previously thought.
Taken together, these findings support a unifying framework: neurodegeneration arises from the breakdown of an interconnected system involving clearance mechanisms, vascular integrity, immune regulation, and synaptic maintenance.
This shift has profound therapeutic implications. Interventions targeting sleep architecture, glymphatic flow, BBB stability, and microglial activation may be more effective than neuron-centric strategies alone. Protecting the system — rather than just the cell — may be the key to altering disease trajectory.
Selected References
DOI: 10.1073/pnas.2516601123
DOI: 10.1126/science.adf6812
DOI: 10.1038/s41591-023-02523-3
DOI: 10.1038/s41593-022-01186-2
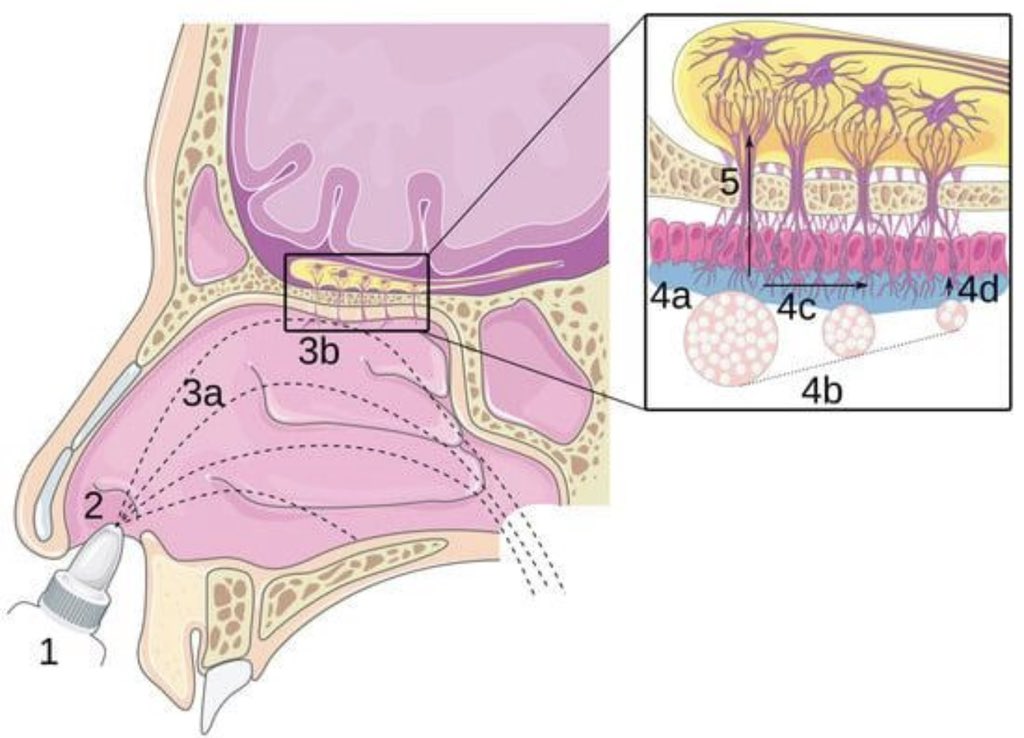
A randomized clinical trial suggests that the antihistamine nasal spray azelastine may reduce the risk of COVID-19 and other respiratory infections when used regularly.
➡️ Participants using the spray were about three times less likely to get COVID-19 compared to placebo and also experienced shorter illness duration.
➡️ The effect is thought to relate to blocking viral entry in the nasal lining and reducing local inflammation, though the exact mechanism is not fully established. 1/
@winslow_la@ActionCovidLong Nous avons constaté que certaines personnes hésitent à participer à un essai contrôlé par placebo par crainte de se retrouver dans le groupe placebo. C'est pourquoi, pour notre essai portant sur la rapamycine à faible dose dans le traitement du #COVIDLong. https://t.co/qv7du3Gv5H
There’s a common argument that we need validated biomarkers before running clinical trials in Long COVID and ME/CFS. I think this is wrong.
First, you don’t need disease-specific biomarkers to track whether a treatment is working. Nearly all clinical trials across medicine rely on symptom-based endpoints. Change in disease status is measured through patient-reported outcomes, functional capacity, and clinical assessments. This is standard. It’s also what we care about, do we care if biomarker X improved if function is still the same? Yes for research, no for approving that drug, which is the purpose of the trial.
Second, biomarkers can help guide which drugs are worth trialing, but their absence isn’t a blocker. We already have plausible mechanistic targets. There’s evidence pointing to mitochondrial dysfunction, so there’s logic to try mitochondrial drugs etc.
Where biomarkers genuinely matter is in identifying responding subgroups, and this is a different problem than having a “disease biomarker.” It’s exploratory, study-specific work: you run omics-based blood profiling on trial participants, or leverage established subgrouping frameworks, and look for signatures that predict who responds and who doesn’t. That’s valuable post analysis, not a prerequisite.
What actually needs to change is the study design itself. Trials in this space need to stop recruiting patients too early in their illness. Enrolling people within the first year creates artificially inflated placebo response rates, because natural improvement is still occurring. Trials also need to run far longer than the one-to-three-month durations that are common now. Six to twelve months is more realistic for detecting meaningful therapeutic effects. In mastocytosis, a disease with mast cell involvement that parallels the theorized mast cell dysfunction in Long COVID and ME/CFS, the most promising therapeutics showed significantly greater improvements at six months and one year compared to short-term endpoints. Combine longer trials and later-stage recruitment with the subgroup biomarker exploration described above, and the clinical trial landscape in these diseases improves dramatically.
The framing that biomarkers must come first risks becoming an excuse for inaction in diseases where patients are waiting for trials that could start today.
The program for the International Long COVID & PAIS Conference 2026 is now online! https://t.co/SHD33RV9dR
Many slots available for oral presentations
Abstract deadline: 15 May 2026
A great way to present your (unpublished) work!
Amsterdam | 26–29 Aug 2026
@ISLCPAIS#longcovid


























![Rainmaker1973's tweet photo. Scientists just found a way to reverse osteoporosis – not just slow it.
Researchers from the University of Leipzig and Shandong University have identified GPR133, a cell-surface receptor that functions as a master regulator of bone-building cells called osteoblasts. When GPR133 is switched on, osteoblasts ramp up activity and dramatically increase bone density.
In mouse experiments, animals engineered without the GPR133 gene developed fragile, porous bones similar to human osteoporosis. However, treating them with a synthetic activator called AP503—a small molecule discovered through computational screening—rapidly strengthened their skeletons. Remarkably, AP503 not only boosted bone mass in healthy mice but also reversed osteoporosis-like damage in older or diseased animals, with even greater effects when paired with physical exercise.
Because GPR133 influences bone density in humans and the underlying molecular pathways are highly conserved across mammals, the findings raise hope for a breakthrough osteoporosis therapy. Unlike existing drugs that merely slow bone loss (and often carry side effects or waning efficacy), a GPR133-activating compound could actively rebuild bone from within.
This approach could be particularly transformative for postmenopausal women, who face the highest risk of debilitating fractures, offering a safer, more effective way to restore skeletal strength.
["The mechanosensitive adhesion G protein-coupled receptor 133 (GPR133/ADGRD1) enhances bone formation." Signal Transduction and Targeted Therapy, 30 June 2025]](https://pbs.twimg.com/media/HHeEaQSWoAABuQ4.png)