From genetic associations to genes: methods, applications, and challenges: Trends in Genetics https://t.co/D5Jy3njNJ1. A review paper from a team effort led by Ting @TingQi2
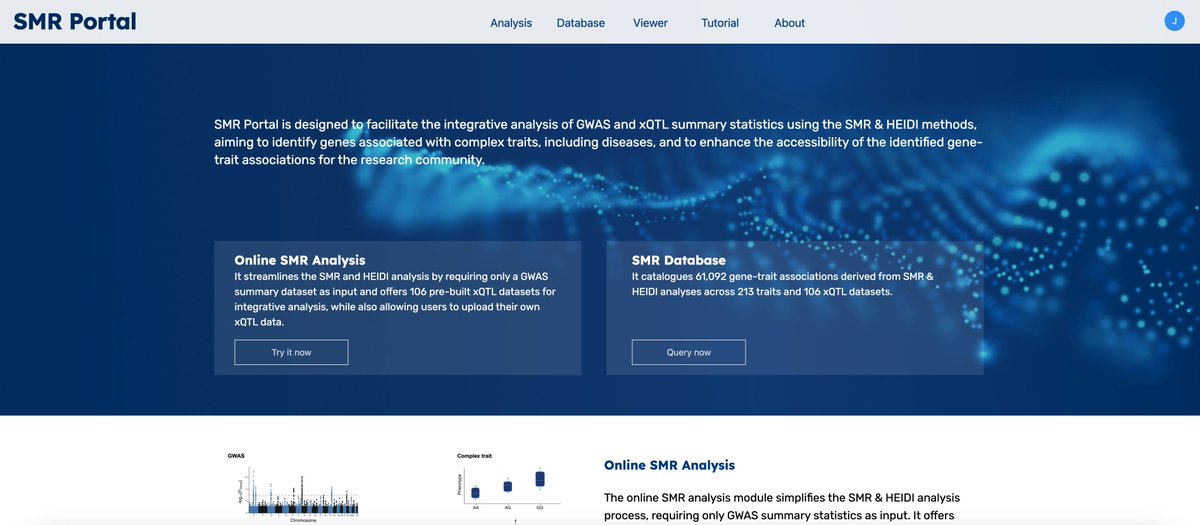
SMR Portal: an online analysis, visualisation, and database platform for mapping complex disease genes. Powered by Westlake FutureGene. Try it out at: https://t.co/bjZiXolaLl. See a tutorial video at: https://t.co/SekxUDfwEg
Estimating the abundances of cells at different states in bulk RNA-seq data. Excellent work from Liyang Song, a second-year PhD student at Westlake University. @liyang_song@Westlake_Uni
The OPERA paper is published in Cell Genomics! The OPERA method integrates GWAS and xQTL summary statistics across multiple omics levels. Findings reveal that 50% of GWAS signals are shared with at least one xQTL signal.
https://t.co/HtW8TXYjPH
@YangWu20@JianZengR@Westlake_Uni
Ting’s paper is out. @TingQi2 Leveraging a powerful new method and large datasets, we established a comprehensive genetic regulation map of RNA splicing, which helped to reveal a distinct and essential role of RNA splicing in common traits and diseases. https://t.co/eXxHNIl3WD
Summary-data-based Bayesian methods (SBayesS and SBayesRS) to estimate the effect size distribution of common variants for human complex traits with respect to the variants' allele frequencies. Great work from Jian Zeng and team! @JianZengR
This study provides evidence for a polygenic architecture of tumor mutational burden and opens an avenue for the use of whole-genome germline genetic variations to stratify cancer patients for immunotherapy. https://t.co/Esx3n0JQd5
In this paper, we show that the discriminative ability of genetic risk score (GRS) in late-onset Alzheimer’s disease (LOAD) prediction is maximised when selecting a small number of SNPs, suggesting LOAD is more oligogenic than polygenic.
fastGWA is ~89 times faster and only requires ~5% of RAM compared to BOLT-LMM-Inf in a data set with n=456,422 and m=8,531,416, and even ~4 times faster than PLINK2.
fastGWA summary statistics for >2000 traits from the UK Biobank are available at https://t.co/aRxdbwMOX0.
Now, we have a robust method to jointly estimate SNP-based heritability, polygenicity, and the relationship between SNP effect and MAF from GWAS summary data. Great work from @JianZengR. Jian Z has also made the software available https://t.co/3GJ6392Rhn.
GTEx V8 public data is now available @GTExPortal. Release V8 includes 49% more RNA-Seq samples from 33% more donors. This release includes splicing QTLs and our new Locus Browser. You can download the Release V8 data here: https://t.co/GaD4mv5CnI #eQTL#sQTL
Detecting GxE by vQTL analysis (without measuring E). 1) Simulation: Levene's test appears to be the most robust vQTL method. UKB data analysis: 2) vQTLs are enriched with known GxE effects; 3) most GWAS loci don’t have a GEI effect. 4) Methods available in OSCA. @huanwei_wang
We've applied fastGWA to common variants in the UKB imputed data for 2,173 traits (n = 456,422) and WES data (n = 46,191) for 2,048 traits. All the summary data are available: https://t.co/YbSsPTT958. An online tool to query the summary statistics: https://t.co/HOrgiEx7V7.
Why it is so hard to predict complex diseases and traits from DNA? Here is a collection of my go-to slides, a short story about the predictive ability of polygenic risk scores (PRS).
Zhang, @jyang1981 and co present OSCA, a method for identifying associations between omics data and complex traits, incorporating the mixed linear model method MOMENT. It has lower FPR than other methods. https://t.co/hCSD2p3rlz