The Molecular Modelling & Engineering Group @uclchemeng, led by @matteoslvlgl. We play with enhanced sampling, nucleation and crystallization processes.
thanks @tiwarylab for the opportunity to talk about our work! credits for the results I have shown go to @nicholasfrancia for the CSP reduction ideas, and twitterless Edgar Olehnovics @UCL_MMEgroup for the hard work on getting flow-based free energy calculations up and running!
The work of Antoniu is now out in @JCIM_JCTC (https://t.co/69hlmGTtYQ) together with an application of MFI to the study of beta scission reactions in explicitly solvated systems led by Francesco Serse in collaboration with Matteo Pelucchi @polimi https://t.co/B7jdm5mDwg
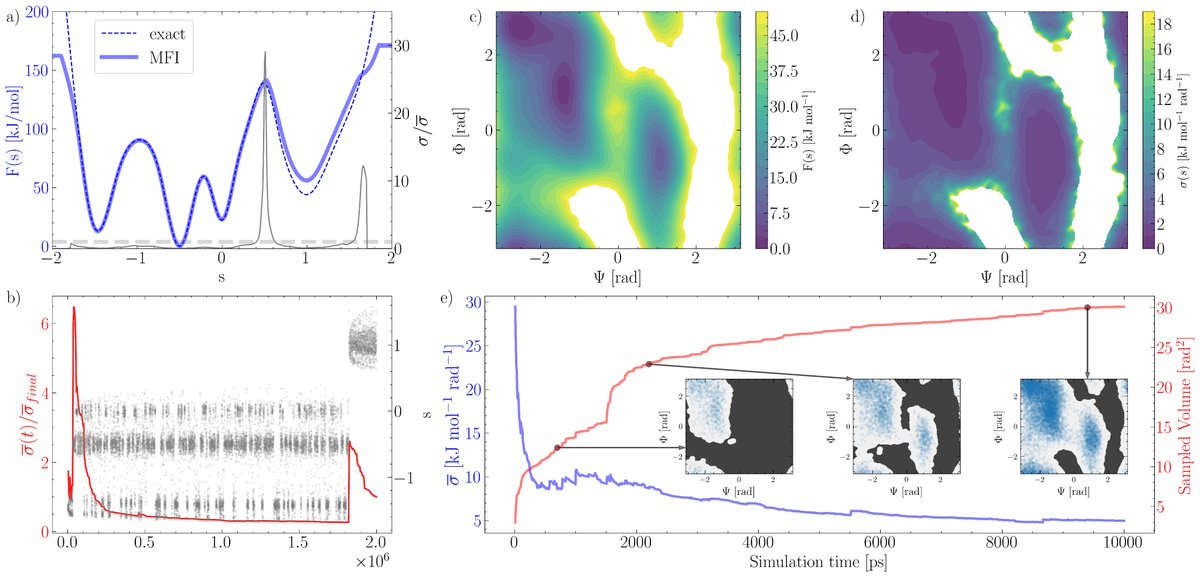
Estimating the convergence of MetaD usually requires a-posteriori analysis. Moreover the sampling obtained while tuning simulation parameters goes unused. Antoniu developed an approach (& code) to tackle these issues: https://t.co/91MtvReEer
@Realworldxstals@uclchemeng
Digitally designing crystallization processes from molecular structure. What does it entail & how close are we? These and other questions are addressed in this (rather extensive) collaborative work led by Chris Burcham @EliLillyandCo : https://t.co/s3TkoEWNn5
A new preprint is out, led by
@FBachtiger!
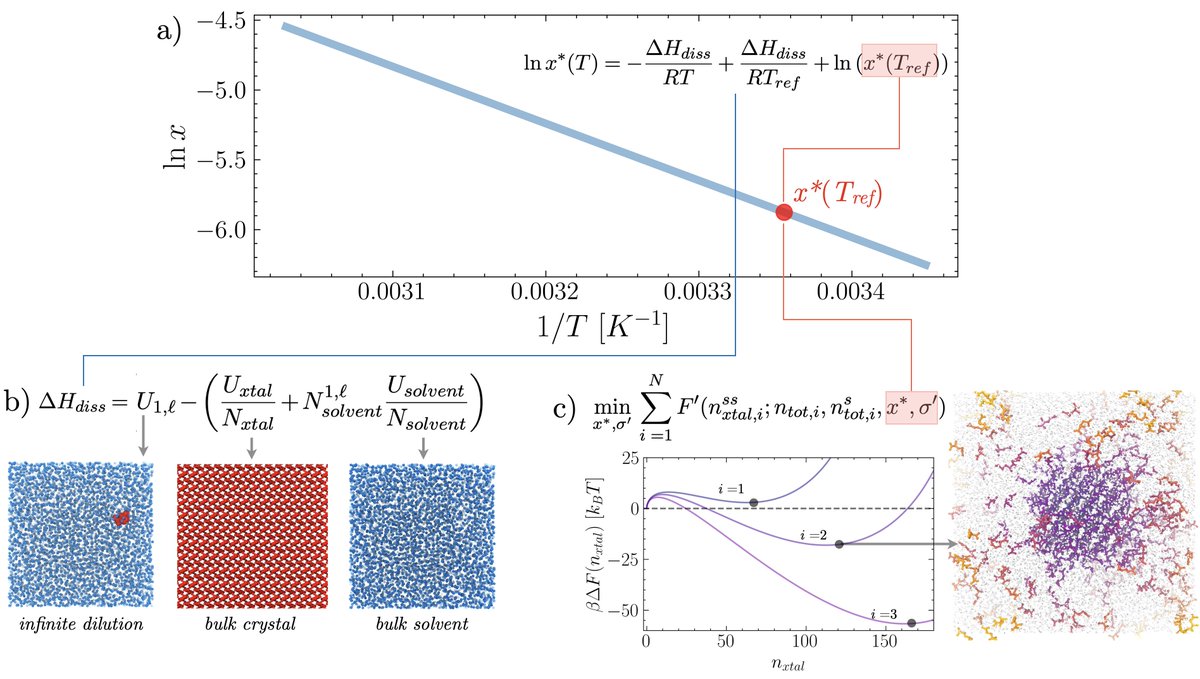
We use thermodynamics of phase transitions in confined volumes to estimate the solubility curve of molecular crystals from a handful of equilibrium MD simulations and assess agreement with exp. data! https://t.co/kEFtxIgnV8
We had the pleasure of welcoming back John Garside (1960-1966) @uclchemeng as we hosted alumni in celebration of 100 years of Chemical Engineering at UCL. Listen to John as he shares fond memories of his time at @UCL. #UCLChemicalEngineeringSince1923
➡️ https://t.co/0xDNa4hyVd
A new preprint & research direction! Led by Edgar and in collaboration with @Michael_Shirts + Ahmad's team @abbvie: Assessing the accuracy and efficiency of free energy differences obtained from reweighted flow-based probabilistic generative models - https://t.co/ichYy4RDp1
We are delighted to announce that Prof Marco Mazzotti @eth_en will be joining @uclchemeng for a Centenary Seminar on "The role of CCS and CDR in reaching net-zero".
Join us on weds, 24 April 2024 @3pm!
#UCLChemicalEngineeringSince1923
➡️ https://t.co/4ozpyik5ix
We have a PhD opening within ht-MATTER - High-throughput modelling of Molecular crystals out-of-equilibrium - @uclchemeng!
If you are interested in molecular simulations, crystal nucleation, & statmech, get in touch!
RTs (RPs?) appreciated!
Details:
https://t.co/U1LuHH13Wz
See below an excellent opportunity to work with me and other researchers at NanoGUNE in a vibrant and international environment in the beautiful city of Donostia-San Sebastián. Contact me if you are interested in applying. The topic will be ML applied to molecular simulation.
Digitally designing crystallization processes from molecular structure. What does it entail & how close are we? These and other questions are addressed in this (rather extensive) collaborative work led by Chris Burcham @EliLillyandCo : https://t.co/s3TkoEWNn5
Estimating the convergence of MetaD usually requires a-posteriori analysis. Moreover the sampling obtained while tuning simulation parameters goes unused. Antoniu developed an approach (& code) to tackle these issues: https://t.co/91MtvReEer
@Realworldxstals@uclchemeng
Explore our new @uclchemeng Centenary publication, offering an overview of our department during the Centenary year 2023-24. Discover the department's rich history and explore our significant accomplishments and milestones spanning the past century.
➡️ https://t.co/cyFPKu5aKk
Funded PhD studentship on ‘scalable design of perovskite solar cells inspired by nature’, (https://t.co/n51R7nZf0x) @uclchemeng via UCL EPSRC DTP 2024/25. Apply here (https://t.co/047wWV7qXO). Home/UK students or international students may apply. Closing date: 8 Jan 2024.
Our review on "Molecular simulation approaches to study crystal nucleation from solutions" is out & open access in WIRES Computational Molecular Science! Thanks to @Sossogroup & the anonymous reviewers for their feedback! @Realworldxstals@uclchemeng
https://t.co/EDaQJgZW6U
Understanding nucleation from solution is crucial in many scientific fields & atomistic simulations can help, but choosing the right method is tricky. In this preprint we review applicability & insights attainable with different MD-based approaches 1/3 https://t.co/OiM05suqpZ