🚀 XMVB Cloud Platform now supports:
• Browser-based molecular structure drawing
• Interactive 3D molecular visualization
• Molecular & valence bond orbital visualization
• Custom orbital colors & display effects
We propose PyMEDA, an open-source, modular Python framework to standardize multiscale embedded EDA workflows for large systems. It has been published in Chin. J. Chem. Phys. 39 (2), 171-181.
#XEDA#CompChem#QuantumComputing
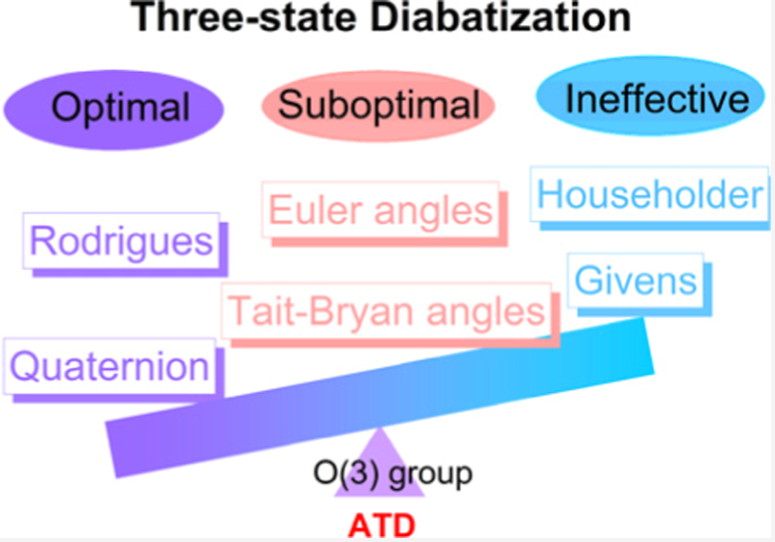
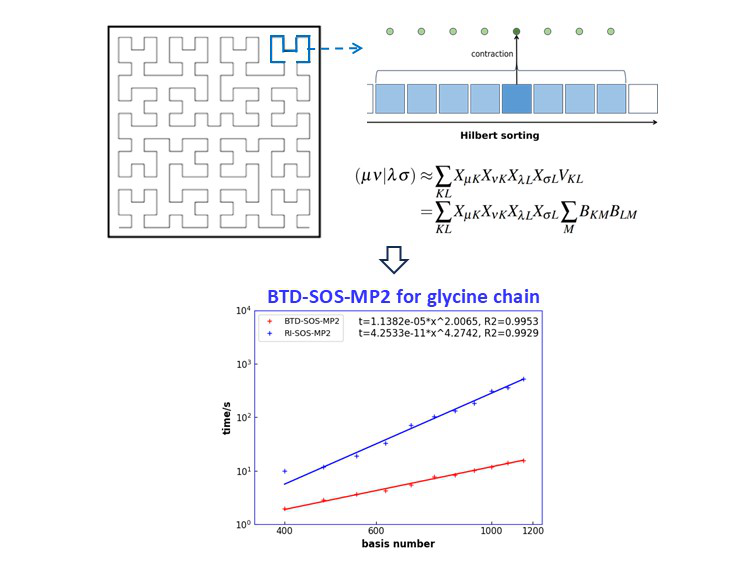
We propose BTD, an efficient, low-scaling algorithm for electron integrals by combining Hilbert space-filling curves with pivoted Cholesky decomposition. It has been published in J. Chem. Phys. 163, 174109 (2025).
#XEDA#CompChem#QuantumComputing
DOI: 10.1063/5.0289370
Recently, we developed BTD-GW, a breakthrough algorithm achieving formal O(N3) scaling that enables high-precision GW calculations for large molecular systems. It has been published in J. Chem. Phys. 164, 144106 (2026). #XEDA#CompChem#QuantumComputing
DOI: 10.1063/5.0319147
🚀 XMVB 4.1 released!
Experience the new generation valence bond computing with a host of new features, available via cloud platform or local installation.
Discover new features: https://t.co/10Q6Co9U9V
XMVB manual:
https://t.co/MafYJfZoGy
#ComputationalChemistry#XMVB
We developed DLVB, the first deep-learning-based framework for VB theory. It predicts VB structure weights without ab initio calculations and provides efficient selected-Cl scheme for compact VB wavefunctions. Published in JCTC. #XMVB#Deeplearning
https://t.co/M9dG4L4zFK
We developed a Block Tensor Decomposition (BTD) algorithm with a formal O(N³) scaling for SOS-MP2 calculations (J. Chem. Phys. 163, 174109, 2025).
#TensorDecomposition#QuantumChemistry#XEDA#CompChem
DOI:10.1063/5.0289370
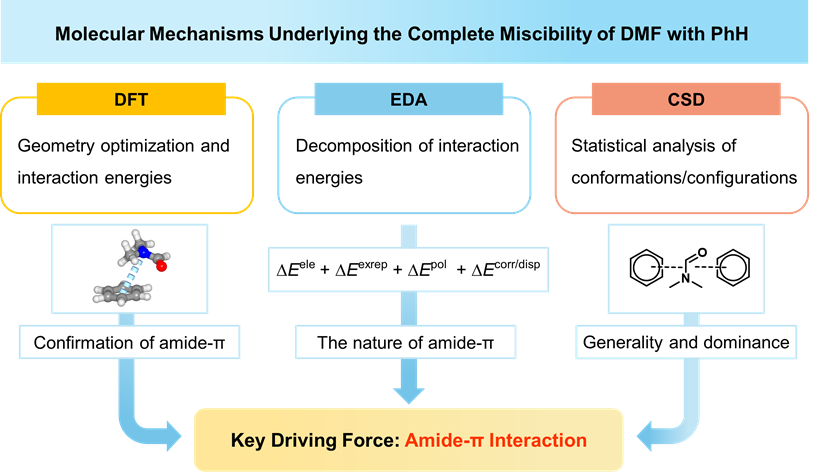
A new study by CanZhong Lu and co-workers uncovers the hidden amide–π stacking that drives the surprising miscibility between polar DMF and nonpolar benzene—bridging electrostatic and dispersion forces and redefining the “like dissolves like” rule.
https://t.co/IFDNboOQ0Z
Online course "Introduction to Valence Bond Theory" is now available. Go to https://t.co/fuC5bp1370 for the course in Chinese and https://t.co/ipJmvxSfto in English.
Recently, we developed a DFT-in-xTB embedded energy decomposition analysis method for analyzing intermole-cular interactions in large systems. It has been published in J. Chem. Phys. 162, 124103 (2025).#XEDA#CompChem#QuantumComputing
DOI:10.1063/5.0258177
Recently, we developed an efficient algorithm for VBSCF energy gradients. By combining XMVB with SHARC, we successfully performed the first ab initio classical VB-based molecular dynamics simulations! #XMVB#Molecular_dynamics#CompChem
DOI: 10.1021/acs.jpca.4c08431
Recently, we published a paper in JCTC. New VB methods (VBSCF(RI), VBSCF(COSX), BOVB(RI)) built on low-rank algorithms achieve 30× speedup, scaling VB to hundreds of atoms. Now implemented in XMVB 4.0.
#XMVB#CompChem#QuantumComputing
DOI: 10.1021/acs.jctc.4c01787
MLatom now offers an easy way to do so: you can leverage the power of the ANI potentials pre-trained on big #data. Check out the video.👇#compchem#machinelearning
https://t.co/5SabxRzJ53
In our latest tutorial we show how MLatom can be used to simulate UV/vis spectra via single-point convolution and nuclear-ensemble approach (NEA). ML can also be used to increase precision of the NEA spectra at reduced cost. #machine_learning#Simulation
https://t.co/Ih4uaj1azZ
We published a #review to explore the current landscape of #molecular quantum chemical #data sets and #databases. It covers key information on more than 40 datasets including the level of theory, size and diversity, methodologies, and availability.
https://t.co/f895utCXiH
We introduce a new approach based on the universal #MachineLearning models of AIQM series targeting the gold-standard coupled-cluster level, going beyond the typical DFT accuracy. Check out the video👇#compchem#dft#IR#Simulation
https://t.co/jgVSvTIkk1