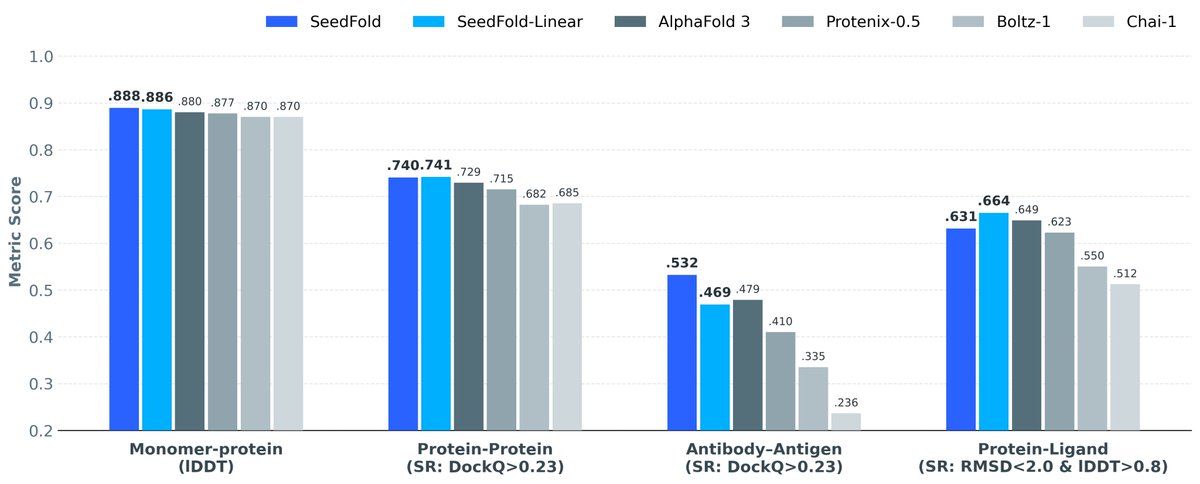
Big news from Boltz - our biggest update yet! 🚀
Today we’re releasing two new state-of-the-art models for protein and small molecule design with extensive wet lab validation and a new API to run all of our models on scalable GPUs wherever you (or your agents) work! 🔥
Meet evedesign: a new open-source ML framework that makes protein design accessible and interoperable.
📢 See our post: https://t.co/lK3Szb3ff8
Protein design models are powerful, but combining them shouldn’t require custom glue code.
✅Combine models for multi-objective optimization
✅Integrate lab-in-the-loop experimental of data
✅100% secure: run on your own infra, no data sharing
Get started building therapeutics & industrial enzymes today 👇
📄Paper: https://t.co/eTOTY6kfZ0
💻Code: https://t.co/Hn3zatn5nc
🌐Webserver: https://t.co/cAM8NVMbL4
Reach out to collaborate: [email protected]
Learning the All-Atom Equilibrium Distribution of Biomolecular Interactions at Scale
1 ByteDance AI Drug Discovery and Anew Therapeutics researchers introduce AnewSampling, the first generative foundation framework that faithfully reproduces molecular dynamics (MD) at the all-atom level for sampling the equilibrium distribution of biomolecular interactions, addressing the high computational cost of traditional MD simulations.
2 AnewSampling leverages a novel quotient-space generative framework to ensure mathematical consistency in its modeling, and is trained on AnewSamplingDB—the largest self-curated database of protein-ligand trajectories to date, containing over 15 million conformations across 10,297 unique protein sequences and 27,979 unique ligand SMILES.
3 The framework builds on an AlphaFold3-like architecture with a stratified hybrid fine-tuning strategy: Low-Rank Adaptation (LoRA) for sequence representation modules and full-parameter fine-tuning for the Diffusion Module, alongside a Cluster-Based Template Guidance mechanism to enforce exhaustive exploration of the equilibrium ensemble.
4 In benchmarking on the ATLAS monomer dataset, AnewSampling outperforms all state-of-the-art generative methods across all 13 evaluation metrics, showing unparalleled accuracy in predicting protein flexibility and distributional accuracy for monomeric systems.
5 For protein-ligand dynamics testing across held-out PDB systems, JACS & Merck industrial datasets and an in-house drug discovery pipeline dataset, AnewSampling achieves statistical alignment with ground-truth MD distributions that far surpasses static predictors and MD-enhanced models like Boltz2, with its generated conformations nearly indistinguishable from MD baselines in key metrics.
6 AnewSampling demonstrates emergent enhanced sampling capabilities beyond conventional MD, successfully navigating high energy barriers to recover coupled ligand and side-chain motions in CDK2 systems (1H1R and 1H1S)—a major challenge for traditional MD that often requires replica-exchange MD (REMD) to achieve.
7 The model accurately captures subtle ligand-induced conformational shifts in congeneric structure-activity relationship (SAR) series, a critical capability for lead optimization in drug discovery, and maintains high fidelity in modeling non-covalent protein-ligand interactions and global protein backbone dynamics across diverse chemical and conformational spaces.
8 The research team proposes a multi-level assessment strategy for generative biomolecular dynamics models, using metrics like Jensen-Shannon (JS) distance for ligand torsion, Wasserstein (WS) distance for protein-ligand interactions and Spearman correlation for Cα RMSF to rigorously validate physical fidelity at the atomic level.
9 AnewSampling offers unprecedented computational efficiency for exploring biomolecular conformational landscapes, enabling integration into research and industrial drug discovery pipelines and driving a shift toward dynamics-aware design of adaptive inhibitors and functional biomolecules.
10 While AnewSampling achieves significant advances, the researchers note current limitations including reliance on structural templates, limited training data for broader biomolecular interaction types (e.g., protein-nucleic acid) and restriction to fixed thermodynamic environments, outlining future work to address these and enable sequence-only equilibrium distribution prediction.
11 AnewSampling and conventional MD are shown to be complementary: MD provides the critical training data for the generative model, while AnewSampling can accelerate MD by generating diverse initial structural candidates that help bypass energy barriers in physical simulations.
📜Paper: https://t.co/bBaP2DPtvu
#AIDrugDiscovery #BiomolecularDynamics #AllAtomModeling #GenerativeAI #ComputationalBiology #MolecularDynamics #ProteinLigandInteractions
Learning the All-Atom Equilibrium Distribution of Biomolecular Interactions at Scale
1 ByteDance AI Drug Discovery and Anew Therapeutics researchers introduce AnewSampling, the first generative foundation framework that faithfully reproduces molecular dynamics (MD) at the all-atom level for sampling the equilibrium distribution of biomolecular interactions, addressing the high computational cost of traditional MD simulations.
2 AnewSampling leverages a novel quotient-space generative framework to ensure mathematical consistency in its modeling, and is trained on AnewSamplingDB—the largest self-curated database of protein-ligand trajectories to date, containing over 15 million conformations across 10,297 unique protein sequences and 27,979 unique ligand SMILES.
3 The framework builds on an AlphaFold3-like architecture with a stratified hybrid fine-tuning strategy: Low-Rank Adaptation (LoRA) for sequence representation modules and full-parameter fine-tuning for the Diffusion Module, alongside a Cluster-Based Template Guidance mechanism to enforce exhaustive exploration of the equilibrium ensemble.
4 In benchmarking on the ATLAS monomer dataset, AnewSampling outperforms all state-of-the-art generative methods across all 13 evaluation metrics, showing unparalleled accuracy in predicting protein flexibility and distributional accuracy for monomeric systems.
5 For protein-ligand dynamics testing across held-out PDB systems, JACS & Merck industrial datasets and an in-house drug discovery pipeline dataset, AnewSampling achieves statistical alignment with ground-truth MD distributions that far surpasses static predictors and MD-enhanced models like Boltz2, with its generated conformations nearly indistinguishable from MD baselines in key metrics.
6 AnewSampling demonstrates emergent enhanced sampling capabilities beyond conventional MD, successfully navigating high energy barriers to recover coupled ligand and side-chain motions in CDK2 systems (1H1R and 1H1S)—a major challenge for traditional MD that often requires replica-exchange MD (REMD) to achieve.
7 The model accurately captures subtle ligand-induced conformational shifts in congeneric structure-activity relationship (SAR) series, a critical capability for lead optimization in drug discovery, and maintains high fidelity in modeling non-covalent protein-ligand interactions and global protein backbone dynamics across diverse chemical and conformational spaces.
8 The research team proposes a multi-level assessment strategy for generative biomolecular dynamics models, using metrics like Jensen-Shannon (JS) distance for ligand torsion, Wasserstein (WS) distance for protein-ligand interactions and Spearman correlation for Cα RMSF to rigorously validate physical fidelity at the atomic level.
9 AnewSampling offers unprecedented computational efficiency for exploring biomolecular conformational landscapes, enabling integration into research and industrial drug discovery pipelines and driving a shift toward dynamics-aware design of adaptive inhibitors and functional biomolecules.
10 While AnewSampling achieves significant advances, the researchers note current limitations including reliance on structural templates, limited training data for broader biomolecular interaction types (e.g., protein-nucleic acid) and restriction to fixed thermodynamic environments, outlining future work to address these and enable sequence-only equilibrium distribution prediction.
11 AnewSampling and conventional MD are shown to be complementary: MD provides the critical training data for the generative model, while AnewSampling can accelerate MD by generating diverse initial structural candidates that help bypass energy barriers in physical simulations.
📜Paper: https://t.co/bBaP2DPtvu
#AIDrugDiscovery #BiomolecularDynamics #AllAtomModeling #GenerativeAI #ComputationalBiology #MolecularDynamics #ProteinLigandInteractions
This is really cool (and wild):
Scientists simulated a complete living cell for the first time. Every molecule, every reaction, from DNA replication to cell division.
The paper (Luthey-Schulten et al., Cell 2026, https://t.co/PXxXWKC8yp), just out today, used JCVI-Syn3A — a synthetic minimal bacterium with fewer than 500 genes. A 3D+time simulation of the full 105-minute cell cycle: DNA replication, protein translation, metabolism, division. Every gene, protein, RNA, and chemical reaction tracked through physical space.
It took years to build. Multiple GPUs. Six days of compute time per run.
And this is the simplest possible cell.
A human cell has ~20,000 genes. It lives in tissue. It interacts with neighbors. It differentiates. It responds to drugs in ways that depend on context we haven't fully measured.
Mechanistic simulation of the minimal cell costs 6 GPU-days for 105 minutes of biology. You cannot scale that to human cells. The complexity isn't 40x harder. It's exponentially harder.
This is why the field pivoted to data-driven models. You can't hand-encode the regulatory wiring of a human hepatocyte. But you can learn it — if you have the right perturbation data collected across enough diverse biological contexts.
The two approaches aren't competing. Papers like this generate the ground truth that future ML models need for validation. But the path to a clinically useful virtual cell runs through foundation models, not through scaling up mechanistic simulation.
Amazing work!
We are launching ML4BioChem club to connect scientists through in-person/online seminars and happy hours to spark new collaborations, and beyond.
Upcoming speakers 👉 : https://t.co/Off5D5Ur0p
Subscribe @ `Email me about events` on website. Will add you to [email protected].
We are launching ML4BioChem club to connect scientists through in-person/online seminars and happy hours to spark new collaborations, and beyond.
Upcoming speakers 👉 : https://t.co/Off5D5Ur0p
Subscribe @ `Email me about events` on website. Will add you to [email protected].
Join us on Monday to discuss "Generative Modeling via Drifting" with the author MingYang Deng!
A one-step generative model trained with an MMD loss.
On Zoom at 9am PT / 12pm ET / 6pm CET: https://t.co/Kew3F4DXag
@silvirouskin Same respond from my dad, also he asked me when i will be a real professor. lol my dad means to be nice, he’s just not familiar with the system.
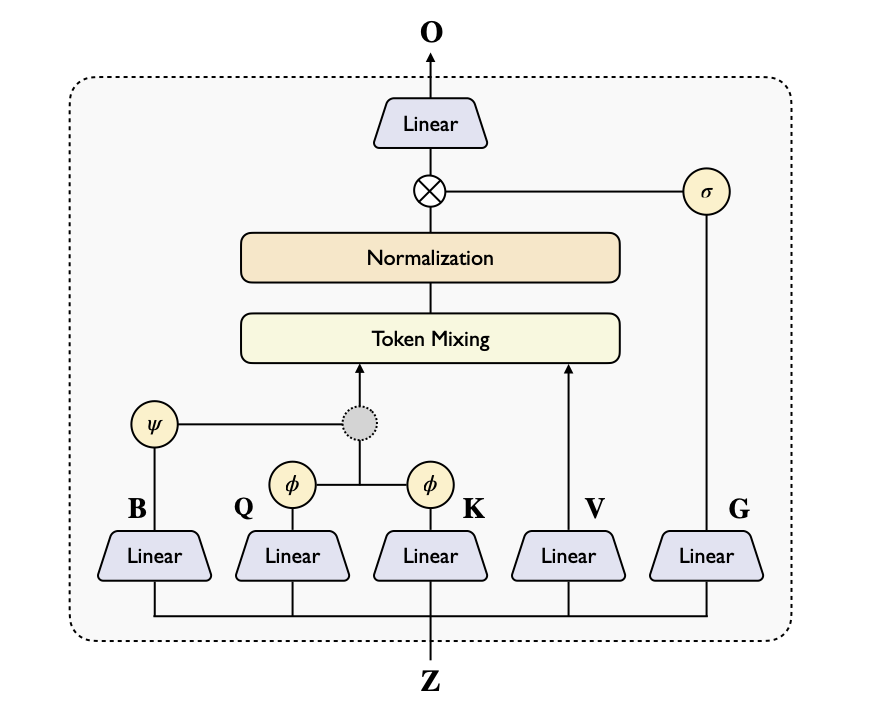
New course: Document AI: From OCR to Agentic Doc Extraction, built with @LandingAI, where I'm executive chairman, and taught by David Park and Andrea Kropp.
Much of the world's data is locked in PDFs, JPEGs, and other documents. This short course shows you how to build agentic workflows that process documents accurately: breaking them into parts, examining each piece carefully, and extracting information through multiple iterations.
Traditional Optical Character Recognition (OCR) captures text but loses context from table headers, chart captions, or reading order of columns. After exploring OCR's limitations, you’ll use LandingAI's Agentic Document Extraction (ADE) framework to process documents. ADE treats pages as visually -- as images -- to parse information and extract fields.
Skills you'll gain:
- Build agents to convert unstructured files into structured Markdown/HTML and JSON
- Use ADE to parse complex data like forms, handwriting, or equations
- Map extracted information to named fields using a specified schema, with bounding boxes for grounding and validation
- Deploy RAG applications with event-driven document processing
Come learn about the best tools for processing documents like financial invoices, medical records, or academic papers intelligently:
https://t.co/PYjgnoaD2K
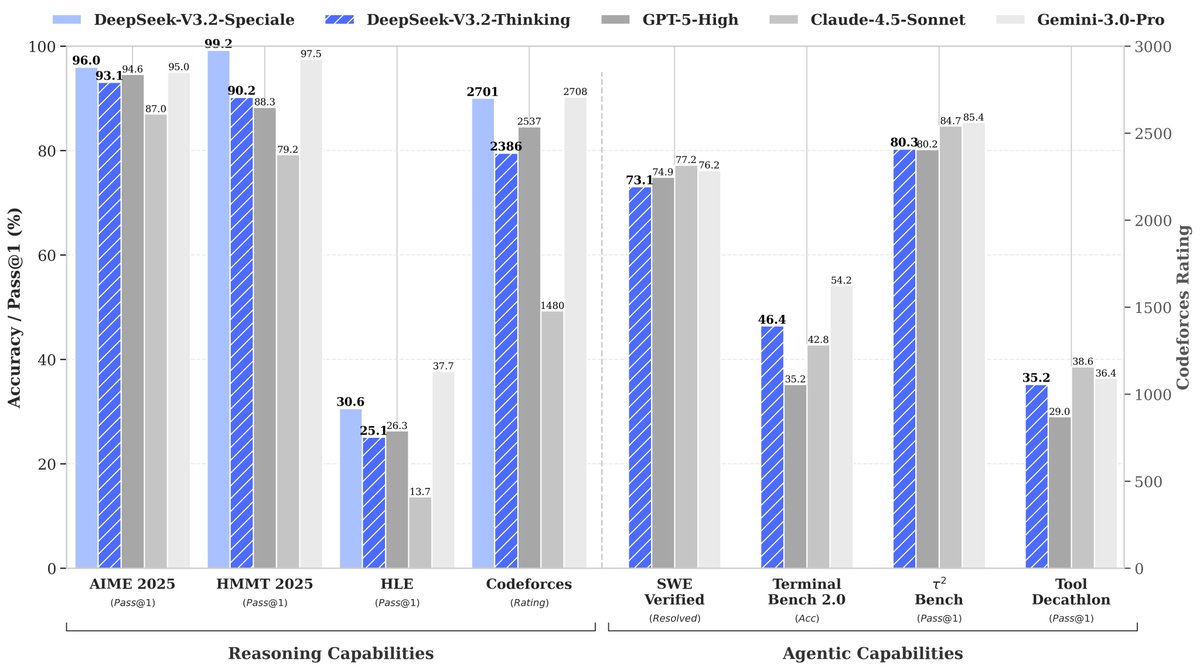
🚀 Launching DeepSeek-V3.2 & DeepSeek-V3.2-Speciale — Reasoning-first models built for agents!
🔹 DeepSeek-V3.2: Official successor to V3.2-Exp. Now live on App, Web & API.
🔹 DeepSeek-V3.2-Speciale: Pushing the boundaries of reasoning capabilities. API-only for now.
📄 Tech report: https://t.co/7EyydyNuG0
1/n
I feel like crap. I didn’t get any grants this year, so I had to let go of my super talented postdoc—someone with a doctorate who’s now unemployed, without benefits or retirement, and still grinding away on a paper... Postdocs are the most unappreciated and undervalued positions at the PhD level-highly qualified, yet paid less than industry techs in Boston (with a BS degree) , forced to work weekends without benefits or job security, and at risk of losing everything if a grant isn't funded. Name another PhD role that's treated with less respect and value by the system.


























![alchemist_an's tweet photo. [ML4BioChem club] 3rd online speaker! @m1nj12, Colin Kalicki! https://t.co/N7ABLyBaMV](https://pbs.twimg.com/media/HJXKzpxXUAE0wty.jpg)
![alchemist_an's tweet photo. [ML4BioChem club] 2nd speaker! https://t.co/Ntp1keMQka](https://pbs.twimg.com/media/HGh5qdNXUAAbo80.jpg)