The abstract submission deadline for Biocontrol26 is coming up Friday May 15th! @bioctrl 🦠🎮
If you are interested in giving a talk or presenting a poster in September at Oxford, you can submit an abstract and see more information on our website:
https://t.co/cgka9KHUii
Happy to share the inaugural editorial written with my fellow chief or senior editors Rodrigo Ledesma-Amaro, PhD & Elia Tomás Pejó.
@LedesmaAmaro@tandfnewsroom
Please submit your manuscripts to Critical Insights in Biotechnology, Taylor & Francis Group
https://t.co/mvXrMV7xAD
Interesting article in Trends in Biotechnology
Influence of titer, rate, yield, and scale on the cost and life-cycle emissions of biomanufacturing: @TrendsinBiotech https://t.co/PuawjJhWMB
📣 Announcing Biological Control Systems 2026, held in-person in Oxford (14-15 Sept) 📣
This event brings together a broad community across #synbio, #control, and #AI/#ML, with contributions from theory to experiments.
Info, abstracts, & registration: https://t.co/abCyWWhPYZ
A non-hyped explainer of the “cell simulation” paper.
The recent study about the “4D” simulation of a minimal cell has been getting a lot of attention on social media. Unfortunately, most posts about it have serious errors. I’ve seen people claim that the model simulates every chemical reaction in the cell, for example, which is not true.
Some biomolecules and reactions *are* tracked individually in the simulation, including proteins and RNA (and ribosomes), and the chromosome. But the simulation does not track individual metabolites (like ATP or glucose), water, nucleotide precursors, lipds, and so on. These "other" molecules are represented, instead, as concentrations (using ordinary differential equations).
But anyway, here goes my quick explanation:
Researchers built a computational model that simulates roughly 100 minutes of biological time, or one cell division, for a single bacterial cell. Each simulation takes 4–6 days to run on two NVIDIA A100 GPUs, and the authors ran it 50 times in replicate. The cell simulation includes some elements of randomness, so each replication attempt leads to a slightly different outcome. When they plotted out these replicates and averaged results, they found that the model could predict a few things without being fitted to experimental data: The simulated cells “divided” every 105 minutes, on average, which matches experimental results; and the mRNA molecules had an average half-life of 3.63 minutes, which is roughly what we’d expect from experiments, too.
The cell they are modeling is called JCVI-syn3A, and it is not a naturally-occurring organism. It’s a bacterium that has been engineered, over many years, to have a small genome. It only has 493 genes (compared to 4,000+ for E. coli), all of which are housed on a single chromosome. The Syn3A cell was made by taking a natural organism, called Mycoplasma mycoides, and then slashing out non-essential genes. Its entire proteome, transcriptome, and metabolism have been studied in depth, which is why it’s being used to build these whole-cell simulations.
The actual *simulation*, though, is not a single thing! Instead, the authors wrote down all the “stuff” that happens inside a cell (transcription! translation! metabolism! lipid biosynthesis!) and decided which type of mathematical model would be best-suited to describe each thing. Some cell processes were modelled deterministically, others had “spatial” elements, and other parts were relatively random.
More specifically, they used four different types of models to build this simulation:
1. A Reaction-Diffusion Master Equation, which was used to model the individual proteins, RNAs, and ribosomes.
2. A Chemical Master Equation, which was used to model things where spatial location doesn’t matter as much (it basically treats the whole cell as one mixed entity); including tRNA charging.
3. Ordinary Differential Equations, which you may be familiar with from Calculus class, were used to model changes in ATP concentration, lipids, and so on.
4. Brownian Dynamics, which simulated the chromosome as a physical chain of beads, where each bead represents 10 base pairs of DNA.
The Reaction-Diffusion Equation works like this: Basically, they chopped up the entire digital cell into a 3D grid of cubes. Each cube measures 10 nanometers on each side. The whole cell is about 500 nanometers across, so there are tens of thousands of cubes in the cell's interior. (This is a useful way to coarse grain the simulation; if the cubes were smaller, the simulation would take much longer to run.)
Each cube is a little box that contains some number of molecules. At every “step” in the simulation, only one of two things can happen to the molecules in each box: Either they react with a molecule in the same box, or they diffuse (“hop”) to an adjacent box. That’s it; the model is just rolling a die for each molecule at each time step in each box, and using those results to decide how each molecule changes over time.
(The reason this spatial model is important is because biology only works if molecules physically bump into each other. And so this spatial grid means that, unlike simpler models, a protein actually has to “diffuse” across boxes in the cell to encounter its reaction partner; only then can it react and do something useful.)
So anyway, each of these models is used to represent a different type of molecule. It’s not like there is a single, all-powerful simulation that they are running here; instead, they’re running these four models together, using a script that synchronizes their results with each other.
The Reaction-Diffusion equation is the main part of the simulation. It takes time steps of 50 microseconds of biological time. Every 12.5 milliseconds of biological time — meaning every 250 RDME steps — the simulation pauses so that the other models can synchronize based on the latest state of the simulation. The Brownian Dynamics part runs on a completely separate GPU, and only updates every four seconds of biological time.
So that's the gist here. But let's also be honest about what this simulation does NOT do:
- It does not include polysomes, which are a cluster of ribosomes that all latch onto a single mRNA and translate at the same time. Polysomes are really common inside of cells, but this simulation assumes that each mRNA can only be translated by one ribosome at a time.
- It does not include polycistronic transcription. In bacteria, genes are often grouped next to each other on the chromosome and thus “transcribed” (or turned into mRNA) all at once, together. The majority of genes in E. coli, for example, are arranged in these operons, and the authors of this paper acknowledge that many Syn3A genes are likely co-transcribed the same way. But the simulation doesn't capture it.
- The authors manually tuned many parameters to get the model to make predictions that more closely resemble experiments. Earlier simulations were waaaayyyyy off from experimental results. For example, they adjusted the ratio of mRNA binding rates to ribosomes versus degradosomes because, in earlier simulations, mRNA was being degraded too quickly, before ribosomes could translate it, causing most proteins to be severely underproduced.
- In the Brownian Dynamics model, the authors added a “fake” 12 pN physical force to push the two daughter chromosomes apart during division, because the real biological mechanism for chromosome partitioning in Syn3A is not known.
- And some other things.
That being said: This model is really cool! I love papers like this! I'm enamored by scientists who choose really difficult problems (like simulating an entire cell) and actually go after it and make progress!
This paper is amazing because it shows us what we are able to simulate well, and what we don't yet understand, and to figure out which experiments we ought to perform to reconcile the two. So instead of framing this paper as "OH MY GOSH SCIENTISTS FIGURED OUT HOW TO SIMULATE AN ENTIRE CELL!" we should frame it as proof that there is still plenty of room at the bottom, many measurements to be made, and many avenues to explore as we seek to understand biology better.
Great Voices article in Cell Systems @CellPressNews by many of my colleagues in Engineering Biology or Synthetic Biology.
What problem do you hope bioengineering or synthetic biology approaches will enable us to tackle in the next decade? https://t.co/fzGjlWyM5w
Prof Luis Serrano will give a welcoming remark at #SynBYSS. Collins’ & Elowitz’s @Nature papers, Jan 2000 started #synbio, along with Becskei & Serrano’s Nature, Jun 2000.
You can meet @NatureComms & @TrendsinBiotech editors
Feb. 23: abstract deadline
https://t.co/lB1rfLp1Bh
What’s in a name? For #Pseudomonas putida KT2440, a lot. A new #mBio commentary argues that renaming this iconic strain to P. alloputida ignores decades of research, identity, & community use. A call for pragmatic, community‑driven microbial nomenclature. https://t.co/XnLhn1otGN
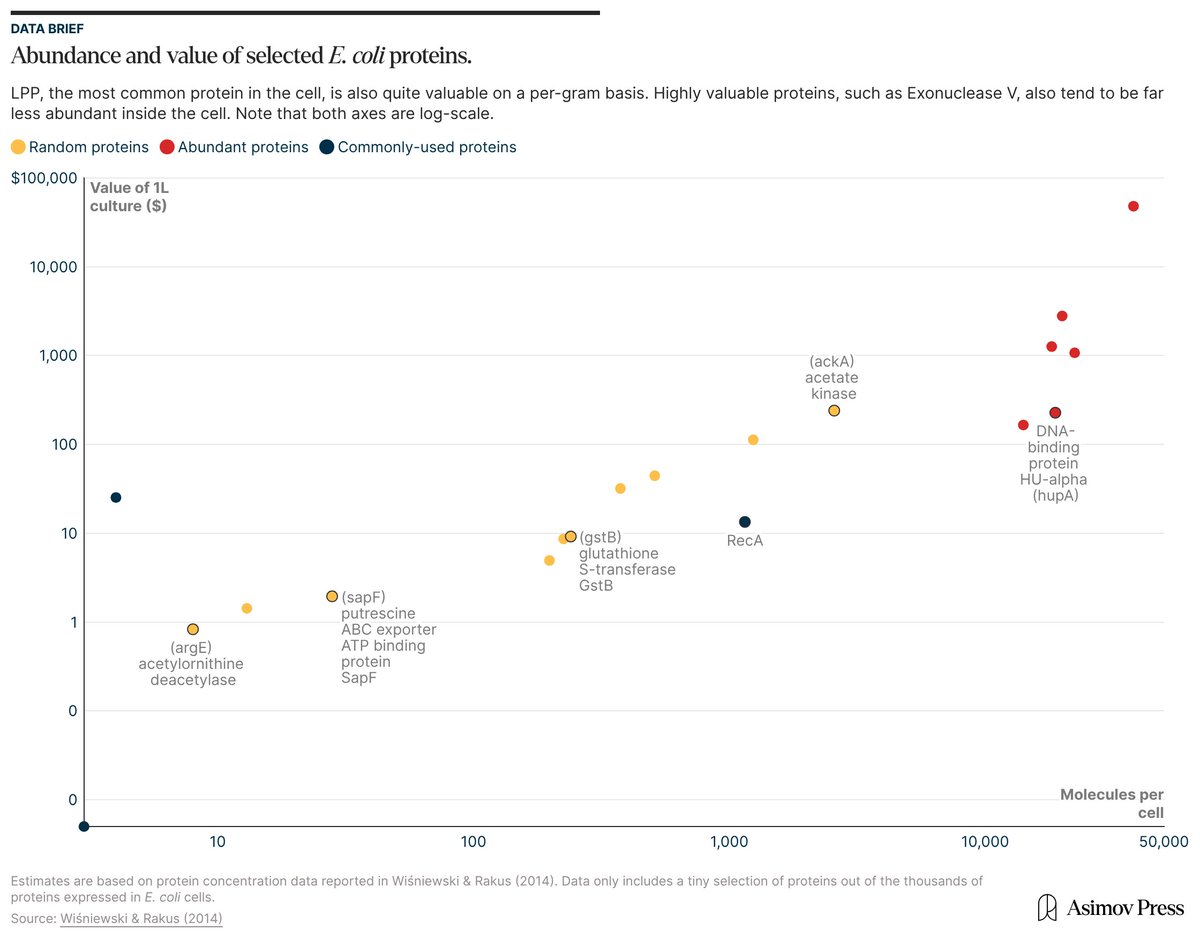
If you took a liter of E. coli cells, isolated all their molecules, and sold them, they'd be worth ~$600,000.
This is a thought experiment, of course; just doing the isolations might cost more than the molecules are worth.
But if you're keen to learn which parts of cells are worth the most money, check out our article: "The Price of E. coli."
https://t.co/QdbW8jofjg
The Synthetic Yeast Genome Project (Sc2.0) set out to redesign and chemically synthesize an entire eukaryotic genome. In 2025, the final synthetic chromosome was completed, marking the culmination of more than a decade of iterative design and international collaboration. How will these findings shape the future of synthetic genomics? https://t.co/QCxxD9wNyr
https://t.co/GNRLhV767m
🖥️🌐Don't miss our online Workshop coming up this Wednesday, Nov 19. Lots of exciting talks, posters, and discussions across timezones!
Check out the full programme at: https://t.co/flia9W21zg
Thrilled to share our new paper in Cell!
We show that phase separation can buffer growth-mediated dilution in synthetic circuits by stabilizing gene expression under dynamic conditions, bringing spatial control into the genetic design toolbox
Proud of our amazing team & collaborators!
https://t.co/8vAoBkceGG
Happy #NationalEngineeringDay! 🚀
This year, we’re launching the AI–Z of Engineering🎉
A digital guide to over 200 current and future engineering roles.
From developing innovative materials to reduce plastic waste🌱to designing VR training games for footballers ⚽and creating smart water systems that waste nothing.💧
👩💻 Crowdsourced from engineers & powered by AI
🌍 Showing how engineering is shaping a better world
👉 Explore the extraordinary breadth and creativity of modern engineering: https://t.co/DBaeAyv7Ou
This article is a homecoming for me.
As a PhD student, I focused on the growth-rate transcriptional regulation in yeast.
Now, ~ 20 years later, we report protein regulation scaling with the growth rates of single cells in mammalian tissues.
https://t.co/qwDYA3v55b