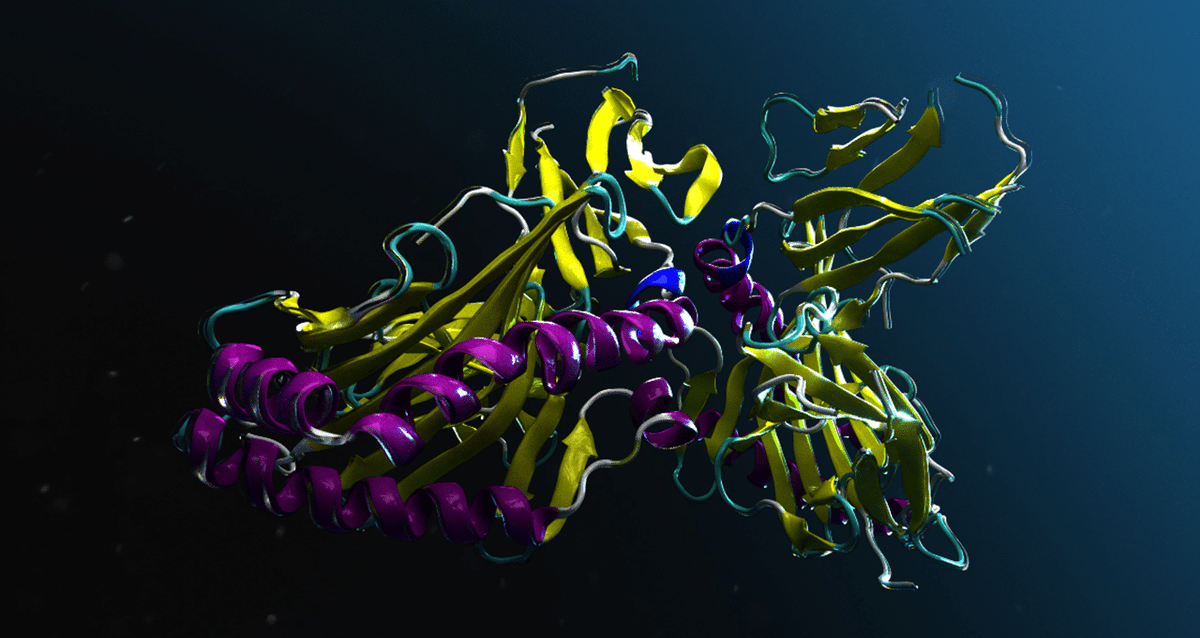
Is ESMFold2 good for PPI discovery? Looks like yes! 🧬
I've previously used literature-based PPI datasets — Y2H reference sets from large-scale yeast-two-hybrid screens in yeast, fly, and human — to test AlphaFold-Multimer for PPI discovery.
I ran a quick test on the fly PPI set with ESMFold2, and it separates positive vs. random pairs remarkably well — and this is the fast model, single-sequence (no MSA), which makes it even more impressive.
We have released the biggest protein data collection on Hugging Face, guys!
We have been working on this for more than 3 weeks now, starting from curating the raw data, doing a lot of filtering, splitting the datasets, sharding them, and doing a lot of analysis. Everything is summarized in our recent blog post.
Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
NEW preprint: Sparse autoencoders were trained on ESM-2 (650M, seq-only) and ESM-3 (1.4B, multimodal) to ask a simple question -- when two protein language models look at the same protein, do they "see" the same biology?
Answer: Yes, mostly
GPT 5.5 is an effective autoresearcher in structural biology!
I've had goal mode running for over 150 hours straight, looking for topologically inspired architectural changes to improve the performance of AlphaFold2.
Performance is strong and improving!
My biggest hot take for today is that everyone who works at the intersection of AI + protein structure should fit an atomic model to density in Coot at least once
Gives you a totally different perspective once you see where the data actually comes from
Graph neural network based hierarchy-aware embeddings of knowledge graphs: Applications to yeast phenotype prediction
1. Kronström et al. introduce Hierarchy-aware GNNs: a framework that couples GNN message passing on heterogeneous KGs with box embeddings constrained by ontology class hierarchies via a semantic loss, so learned representations better respect domain semantics.
2. Key idea: treat each GNN layer’s node output as latent variables that parameterise axis-aligned boxes; then enforce ontology constraints (e.g., subClassOf as geometric containment, and optional disjointness as non-overlap) using semantic losses during end-to-end training.
3. The KG is built for Saccharomyces cerevisiae by integrating curated resources (SGD, GO, APO, ChEBI, INO, MI, RO, BioCyc) and rewriting ABox facts into a TBox-style form, enabling a uniform representation of individuals/classes and relations as axioms usable for both GNN computation and semantic constraints.
4. The model targets quantitative phenotype prediction: predicting fitness (cell growth) for double gene deletions (digenic knockouts) as an edge-level regression problem over gene nodes, trained on 10,085,183 gene-pair measurements (standard condition 30°C), with careful removal of overlapping interaction edges from the KG to avoid leakage.
5. Architecture details: a heterogeneous GraphSAGE-style GNN (relation-specific modules per source-edge-target type) produces gene embeddings; deleted gene pairs are combined with a symmetric operator (best: element-wise product) and passed to an MLP regressor. Domains are embedded separately (8 ontology-aligned domains) to improve stability and efficiency.
6. Main performance result (10-fold CV, split by genes to prevent overlap between train/validation genes): a task-only GNN reaches mean R2=0.348; adding subClassOf links directly to the KG yields R2=0.350; using pretrained box embeddings as priors improves to R2=0.360; adding semantic loss improves further to R2=0.368 (overlap loss) and best R2=0.377 (distance-based loss).
7. Baselines: LightGBM on sparse phenotype instantiations achieves R2=0.211; LightGBM on a 64-dim ComplEx KG embedding achieves R2=0.191. This supports the claim that end-to-end heterogeneous GNN embeddings, especially with hierarchy-aware constraints, extract more predictive signal from the KG than these feature/embedding baselines.
8. Generalisation test: models trained on digenic deletions are evaluated on trigenic deletion fitness (15,095 triples). Using a 3-way element-wise product, performance reaches R2=0.380 with box priors, and R2=0.415 with distance-based semantic loss, indicating transfer beyond the original pairwise setting.
9. Interpretability-to-experiment loop: using gradient-based attribution over KG-linked traits, they score co-occurring relations important for predictions, generating hypotheses about interacting phenotypes. A selected, lab-feasible hypothesis linked inositol utilisation with NaCl (osmotic) stress resistance; an automated-lab perturbation experiment found a significant interaction, with inositol supplementation rescuing growth under salt stress.
10. Additional contributions: (i) shows how to learn low-dimensional GNN-driven box embeddings without a prediction task (pure semantic training), comparing distance vs overlap losses and visualising 2D molecular-function embeddings; (ii) explores using embedding changes under proposed edge additions as a way to rank KG revisions, finding some relation types show distinguishable rank distributions versus random edges, though effects can be small and variable.
📜Paper: https://t.co/SzDrahlftO
#KnowledgeGraphs #GraphNeuralNetworks #Ontology #RepresentationLearning #BoxEmbeddings #ComputationalBiology #SystemsBiology #Yeast #PhenotypePrediction #ExplainableAI
NEW: today OpenBind ‘comes out of stealth’ so to speak with their first data dump of ~900 novel protein-ligand structures - most with paired affinities
This represents a meaningful %-age increase in all of humanities P-L data in the PDB collected in the last 50 years
More👇
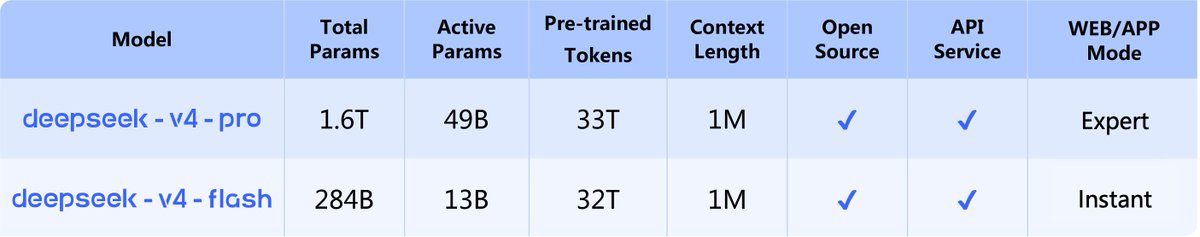
🚀 DeepSeek-V4 Preview is officially live & open-sourced! Welcome to the era of cost-effective 1M context length.
🔹 DeepSeek-V4-Pro: 1.6T total / 49B active params. Performance rivaling the world's top closed-source models.
🔹 DeepSeek-V4-Flash: 284B total / 13B active params. Your fast, efficient, and economical choice.
Try it now at https://t.co/GCdiMzk1Dl via Expert Mode / Instant Mode. API is updated & available today!
📄 Tech Report: https://t.co/drlDrxkYtp
🤗 Open Weights: https://t.co/T13Y8i7SDM
1/n
When your experiment has stages, your optimizer should too
Most real experiments in chemistry and materials science are not single black-box evaluations. They are cascades: you deposit a layer, measure its quality, then fabricate the device and measure efficiency. You synthesize a molecule, compute a proxy property, then run the expensive assay. Standard Bayesian optimization ignores this structure entirely, treating the whole pipeline as a monolithic input-output function.
Torresi and Friederich introduce Multi-Stage Bayesian Optimisation (MSBO), a framework that explicitly models these cascade dependencies and makes decisions at each stage based on intermediate measurements — what they call proxy measurements.
The architecture has three core ideas working together. First, a cascade of independent Gaussian processes, one per stage, propagates uncertainty through the full pipeline via Monte Carlo sampling. Second, a nested acquisition function evaluates the expected utility of a decision at stage i by integrating forward over the probabilistic outcomes of all downstream stages. Third, an inventory system enables resumable sampling: the algorithm can pause a candidate at any intermediate stage, continue it later, or discard it early if the proxy measurement signals low utility — without re-running preceding steps.
The results are systematic. Across nine combinations of stage complexity in synthetic benchmarks, MSBO consistently outperforms both standard BO and BOFN. In real-world tasks — optimizing HOMO/LUMO levels from the QM9 dataset, aqueous solubility, and hydration free energy — MSBO identifies top-1% molecules using roughly half the budget of standard BO. For LUMO optimization, it reaches the top 0.01% of candidates, an order of magnitude deeper than the baseline. Crucially, even when proxy measurements are weakly correlated with the final objective, MSBO still outperforms standard approaches.
For R&D teams running multi-step experimental pipelines — whether in drug discovery, catalyst development, or materials synthesis — this directly addresses one of the most persistent inefficiencies: the tendency to commit expensive downstream resources to candidates that could have been screened out cheaply at an earlier stage. MSBO provides a principled, data-efficient way to build that funnel without requiring pre-defined selection thresholds, replacing human-designed filters with learned, adaptive decision-making across the full cascade.
Paper: Torresi and Friederich, Digital Discovery (2026) — CC BY 3.0 | https://t.co/rviA9wCeMO
karpathy is showing one of the simplest AI architectures that actually works..
dump research into a folder, let the model organise it into a wiki, ask questions, then file the answers back in.
the real insight is the loop...every query makes the wiki better. it compounds.. now thats a second brain building itself.
i think this is so good for agents if applied right
instead of pulling from shared memory every session, they build a living knowledge base that stays.
your coordinator is not just coordinating tasks anymore.. it is maintaining institutional knowledge so every execution adds something back to the base.
the bigger implication is crazy tho.
agents that own their own knowledge layer do not need infinite context windows, they need good file organisation and the ability to read their own indexes.
way cheaper, way more scalable, and way more inspectable than stuffing everything into one giant prompt.
ROCKET 🚀 inference-time optimization of AlphaFold to fit structural data is published! https://t.co/uogMbJZMXs
Since our preprint, we’ve pushed it to regimes where other methods break: low resolution, weak signal, real experimental edge cases. Here’s what we learned: 1/15
Boston biotech has been running the same playbook for years and everyone in the ecosystem knows it.
Early-stage companies are built less on validated biology and more on signaling: a splashy Nature or Science paper, a thin patent scaffold, and the reputational gravity of well-networked academic founders. That combination is often enough to unlock large funding rounds.
The problem is that high-impact publication has become a proxy for truth. It isn’t. It’s a selection mechanism for novelty and narrative.
The result is predictable:
– groupthink gets reinforced
– weak or irreproducible findings persist for years
– dissent is disincentivized
– hype substitutes for validation
In many cases, the goal is not to rigorously test whether an idea is correct, it’s to create enough mystique that it feels important. That perception alone can carry a company surprisingly far.
So it’s not surprising to see the same voices recycled across boards and advisory roles—people who helped build and legitimize this model in the first place.
📢 We’re launching Proteina-Complexa — and after the Jensen keynote mention, we definitely had to post this thread now ;)
Atomistic binder design with generative pretraining + test-time compute, plus large-scale wet-lab validation.
Project page: https://t.co/aT8Lz2VhSJ
🧵 1/n
There might be an interesting, tractable project to fine-tune plms on extremophiles from e.g., https://t.co/o6JVo8vK7Q
There are thermostability plms, but there should also probably be pH, radiation, salinity, etc. I searched around a bit but didn't find this...
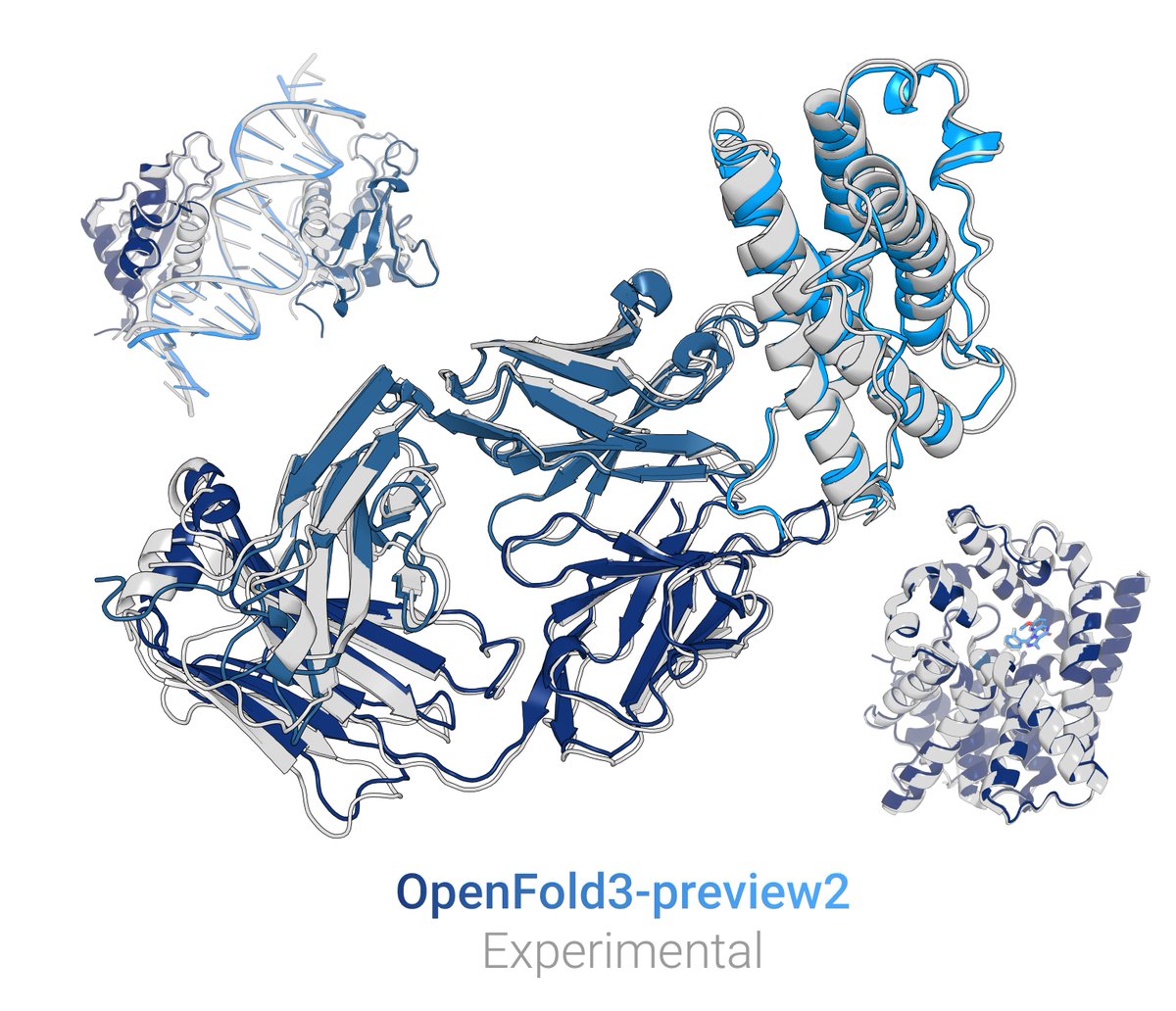
New OpenFold3 preview out! (OF3p2)
It closes the gap to AlphaFold3 for most modalities.
Most critically, we're releasing everything, including training sets & configs, making OF3p2 the only current AF3-based model that is functionally trainable & reproducible from scratch🧵1/9