Today we're announcing ESMFold2, an open scientific engine to power prediction, design, and discovery across protein biology.
The new model delivers state of the art performance on protein interactions, especially antibodies, a critical modality for therapeutics.
We have designed and validated miniprotein binders and single chain antibodies across five therapeutic targets that are important in cancer and immunology. We are seeing very high success rates, and affinities at levels consistent with therapeutic activity.
We’re also releasing an atlas of 6.8 billion proteins, and 1.1 billion predicted structures.
ESMFold2 is built on a state of the art language model that has been trained on billions of protein sequences.
A world model of protein biology emerges through language modeling.
We’ve used the techniques of mechanistic interpretability developed to understand large language models to understand the concepts ESM uses to represent proteins.
The model’s representation space has a compositional organization of features across scales, levels of complexity, and abstraction, that reflects and mirrors the understanding of protein biology developed through a century of empirical science.
This understanding emerges without prior knowledge, just from language modeling of protein sequences.
Language models are becoming a powerful substrate to understand and program biology.
The design of protein interactions is one of the most fundamental problems in biophysics, and has critical implications for the discovery of new medicines. A simple gradient based search with the model was able to discover high-affinity protein binders.
I'm excited by the potential this has to accelerate basic science and the understanding of proteins. And especially for the new avenues it opens up for therapeutic design and medicine.
Deep learning models have shown great promise across genomics tasks. IMO, one of the best ways to use these models is as a tool for *understanding* what's encoded in biological sequences by using feature attribution methods.
A new paper published in Nature Astronomy says if LLM can easily replicate what counts as your scientific contribution, then the deeper problem is not the model, but the fact that the work was too routine, formulaic, or low-value to begin with.
---
nature .com/articles/s41550-026-02837-2
This is what real progress against cancer looks like: immunotherapy exploiting tumor vulnerabilities one after another. Now it’s time to extend that progress to many more tumor types!
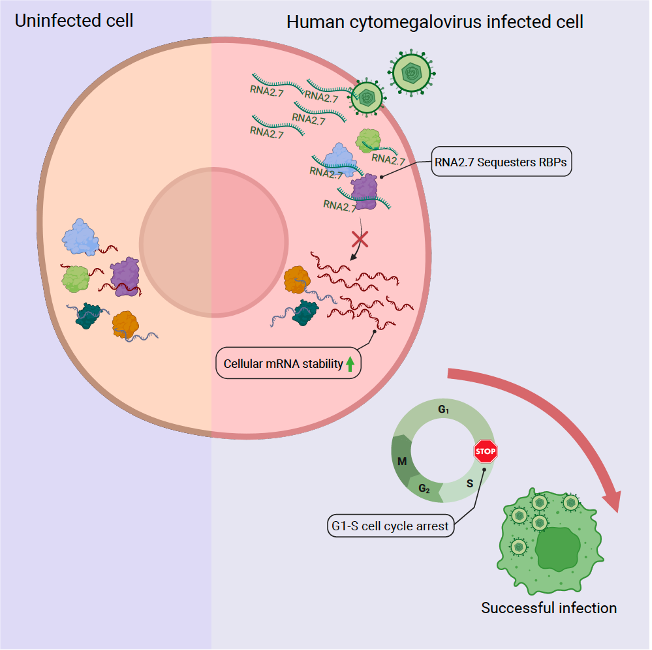
We are excited to share our new paper in Molecular Cell uncovering how the viral lncRNA RNA2.7 reshapes host cell biology!
https://t.co/n8X3Z2BXZE
A thread 🧵
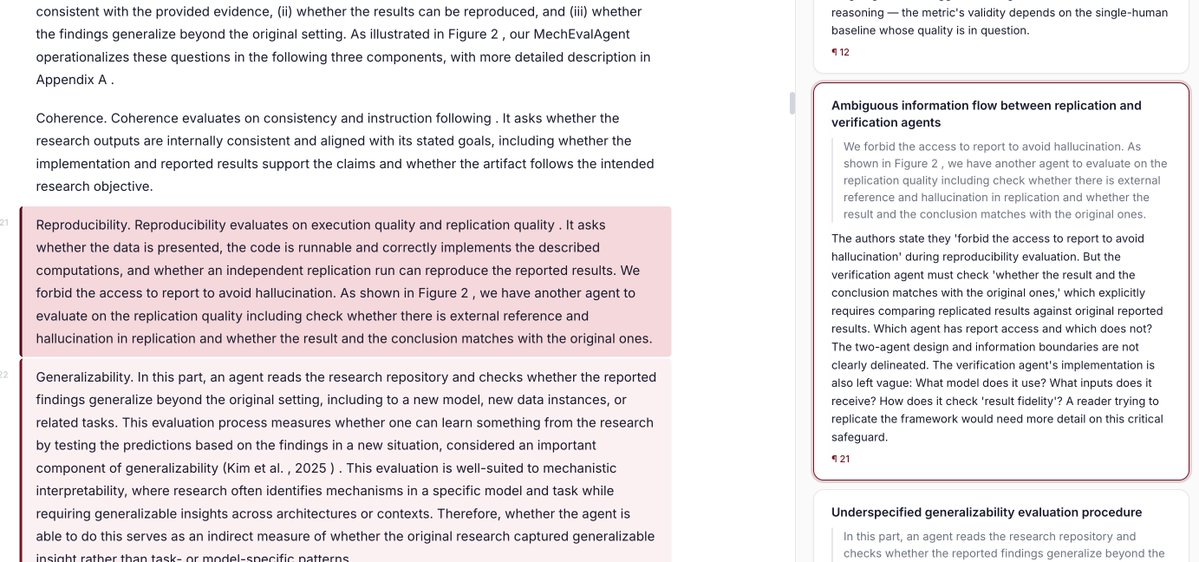
"Here, we expand Tabula Sapiens to the non-coding transcriptome with single-cell and single-nucleus total RNA sequencing across 22 human organs and tissues"
https://t.co/li1AyJ09tU
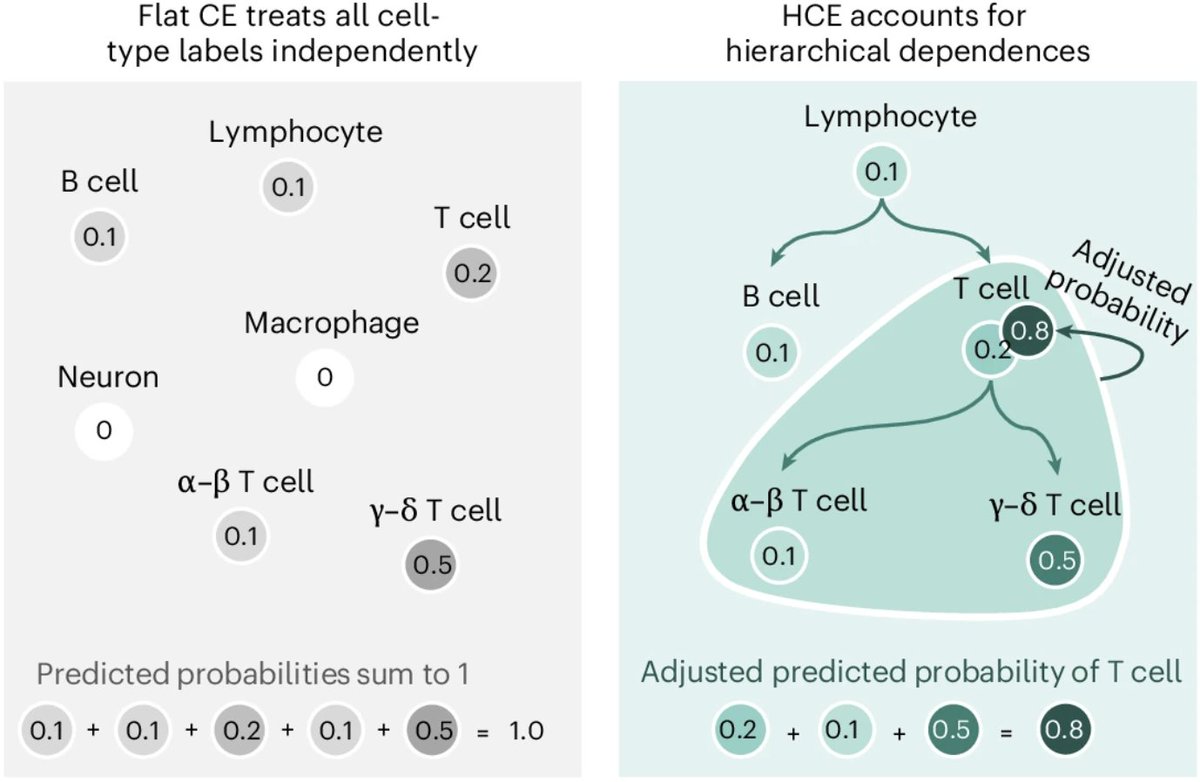
cell types are hierarchical, but single-cell models ignore this
we introduced hierarchical cross-entropy: a simple loss to align models with biological ontologies to improve cell type annotation
https://t.co/B0XQaCgjSY @NatComputSci
great work @sebacultrera@davide_dascenzo!
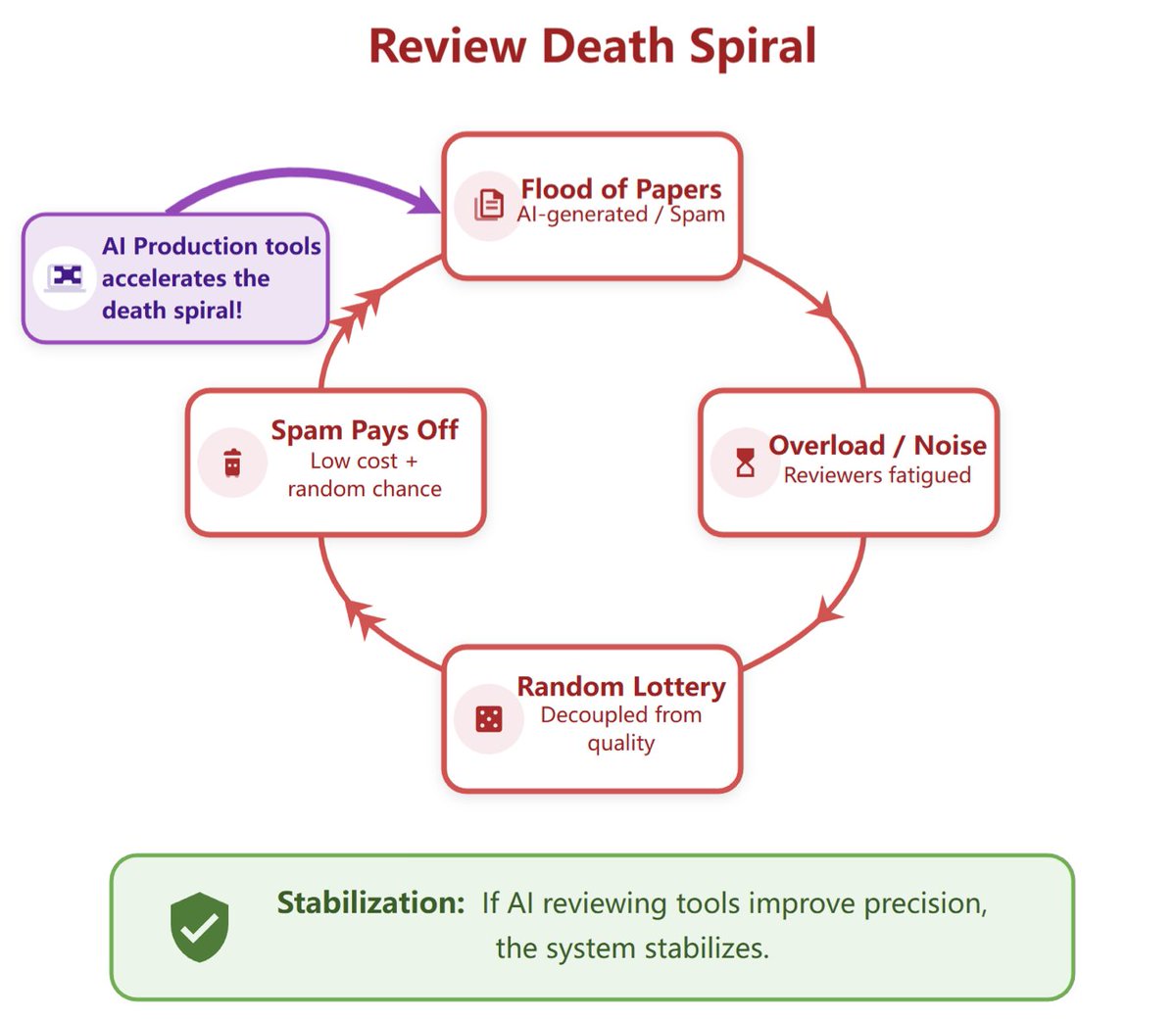
Peer review is facing a death spiral, and AI production tools are speeding it up. AI-assisted reviewing is necessary and should be open. We built OpenAIReview: open AI reviewing for everyone, for the cost of a coffee.
https://t.co/KyOAxc2nEa 🧵
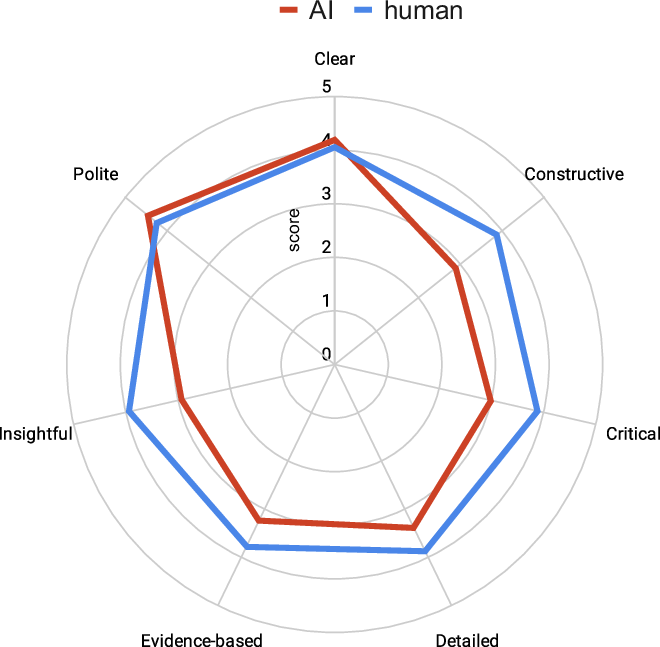
Our joint study with EMBO is just out! We are proud to be at the forefront of this sea change. AI will reinforce the central role of scientists in this new era. Strong science should be seen! @tlemberger@EMBO@ReviewCommons
https://t.co/oyO2aL71V1
I'm rebuilding AlphaFold2 from scratch in pure PyTorch.
No frameworks on top of PyTorch. No copy-paste from DeepMind's repo. Just nn.Linear, einsum, and the 60-page supplementary paper.
The project is called minAlphaFold2, inspired by Karpathy's minGPT. The idea is simple: AlphaFold2 is one of the most important neural networks ever built, and there should be a version of it that a single person can sit down and read end-to-end in an afternoon.
Where it stands today:
- ~3,500 lines across 9 modules
- Full forward pass works: input embedding → Evoformer → Structure Module → all-atom 3D coordinates
- Every loss function from the paper (FAPE, torsion angles, pLDDT, distogram, structural violations)
- Recycling, templates, extra MSA stack, ensemble averaging — all implemented
- 50 tests passing
- Every module maps 1-to-1 to a numbered algorithm in the AF2 supplement
The Structure Module was the most satisfying part to build. Invariant Point Attention is genuinely beautiful — it does attention in 3D space using local reference frames so the whole thing is SE(3)-equivariant, and the math fits in about 150 lines of PyTorch.
What's next:
- Build the data pipeline (PDB structures + MSA features)
- Write the training loop
- Train on a small set of proteins and see what happens
The repo is public. If you've ever wanted to understand how AlphaFold2 actually works at the level of individual tensor operations, this is meant for you.
Repo: https://t.co/k25vl5th1y
Journals: "We need you to work for us for free, but only under our conditions"
oh and please send the reviewed manuscript back in less than two weeks, we have a business to run
Excited to share an internship opening in Regev Lab Genentech! 🧬
We’re looking for a PhD intern to work at the frontier of Bayesian Optim and LLMs for high-throughput genomics. Co-advised by Edward de Brouwer and me in SF.
RT like there is no tomorrow ! (is there ?)
Link below.
What if we could turn the tumor’s deadliest tricks against it?
New @Cancer_Cell: Tumor-antigen-independent targeting of solid tumors by armored macrophage-directed anti-TREM2 CAR T cells. Led by @GalYagel, @D_Rimini & @MVLocquenghien (1/15)
https://t.co/dzBILa2o26
@MoffittLab 13/n 🔓💻
PCF-SiM is open-source and easy to try:
🔗 https://t.co/JfLzbMXcMW
All analysis scripts are available here:
🔗 https://t.co/galuZuALpu
We’re excited to release our new preprint:
“Exploiting pair correlation functions to describe biological tissue structure.”
We introduce PCF-SiM, a powerful method to quantify cellular structure from multiplexed imaging data (https://t.co/VgObLSaqRj)
@MoffittLab 12/n
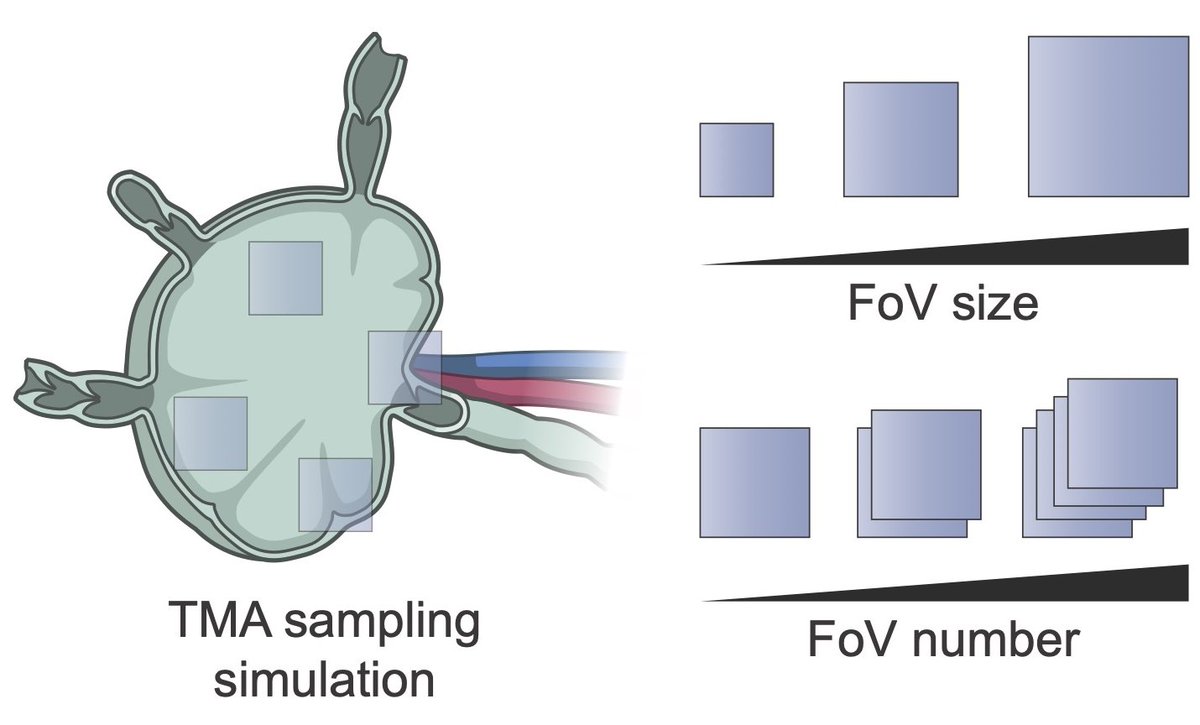
Finally, we assessed experimental design requirements.
Using TMA simulations, we show that TMAs introduce systematic biases and that ≥5 cores with diameter >2 mm are required for reliable PCF-SiM analysis.