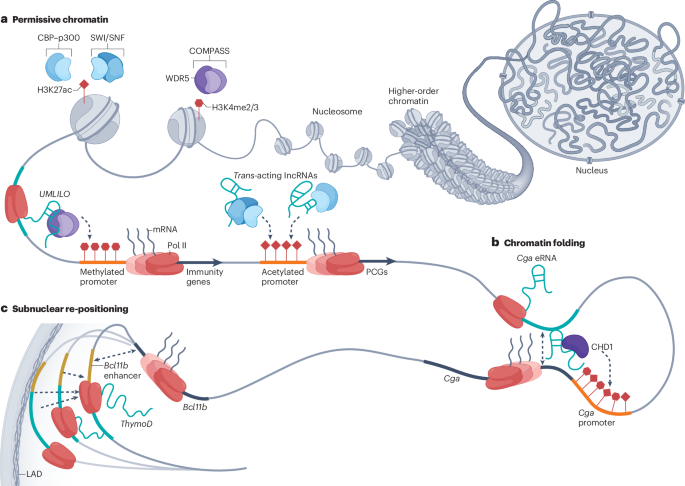
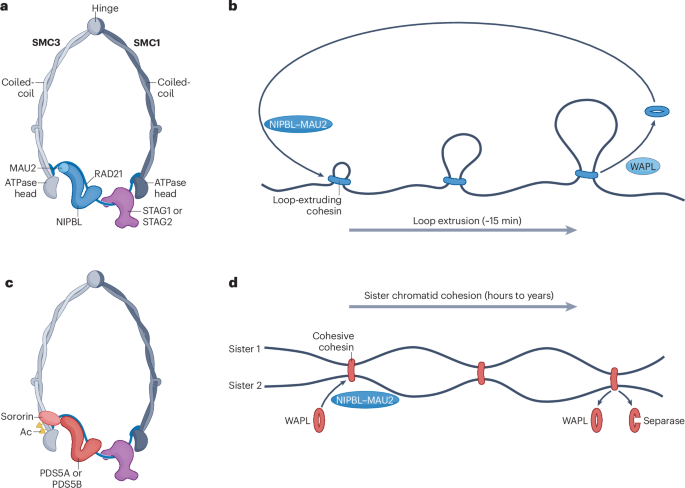
Together with UC Berkeley we are announcing the laser phase plate - a breakthrough in atomic resolution imaging. This is the brightest continuous wave laser in the world, 100 million times the intensity of the surface of the sun.
Phase contrast plays an important role in microscopy, but it was thought close to impossible for electron microscopy, where it would require interfering with an electron beam. Holger Mueller and Robert Glaeser proposed exactly this using a standing wave laser. It has taken over 15 years to make this a reality. Biohub partnered with UC Berkeley and Mueller to support this work and to engineer and build the technology.
Contrast has been the critical barrier to achieving atomic resolution imaging of the cell. In cryo-electron tomography, a cellular imaging technology that uses electron microscopy, the low contrast makes it impossible to resolve anything but the largest proteins within their cellular context. The laser phase plate removes that barrier.
With advances in AI this breakthrough in contrast will start to open up a new frontier in structural biology, that will allow us to see the molecular machines of the cell, and how they assemble into far more complex and dynamic systems, and understand how they work.
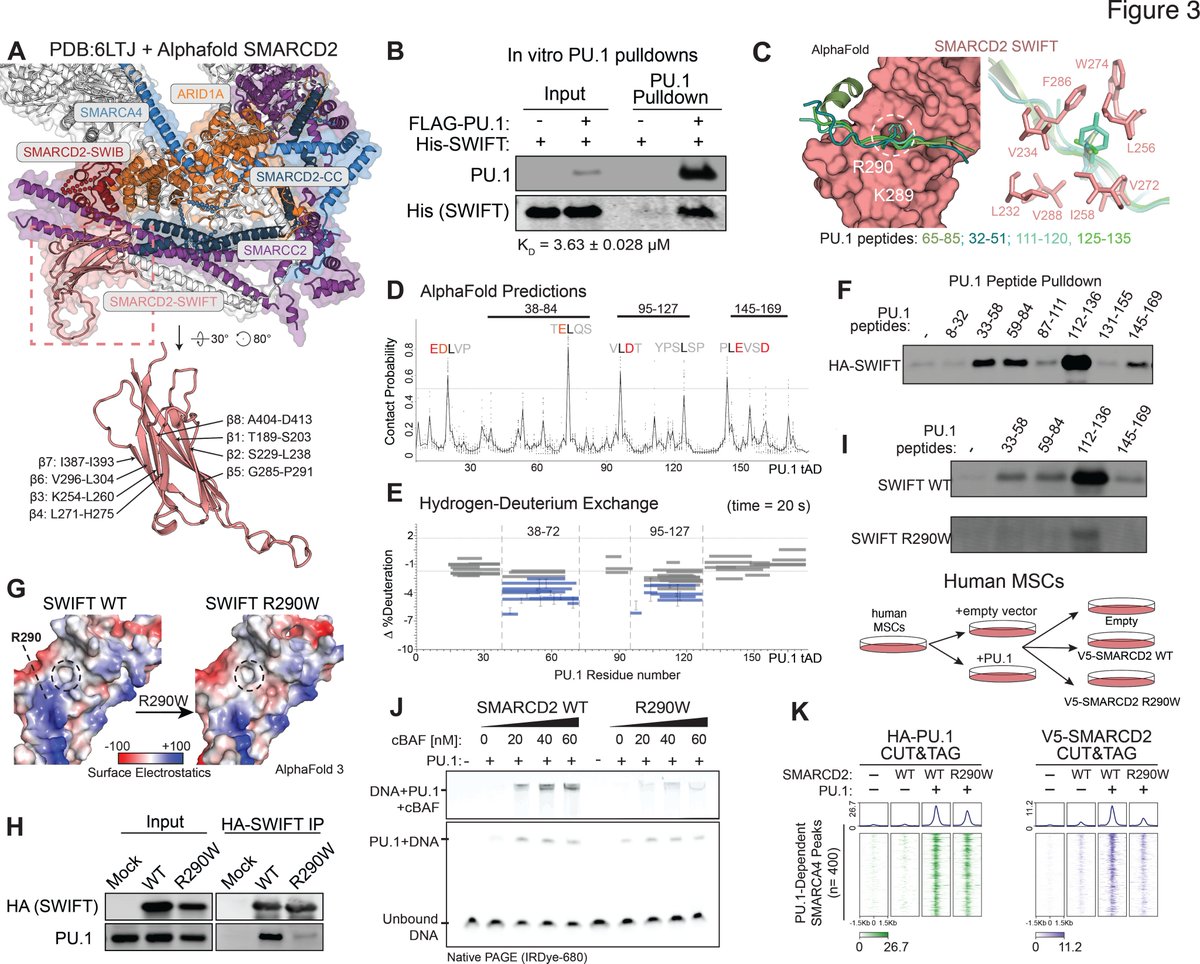
Excited to share our work uncovering SNOR, a ribosome-associated factor that promotes translation restart after dormancy published in Nature!! This was, as usual, a wonderful collaborative effort between our group @UVA and @simonemattei@EMBLHeidelberg
https://t.co/YKDCLFowvC
ROCKET makes AlphaFold context-aware. We iteratively steer prediction to agree with experiment (cryo-EM, crystallography) at inference time, no retraining. Structure determination becomes a search where ML priors and experiment productively combine and inform each other. 2/15
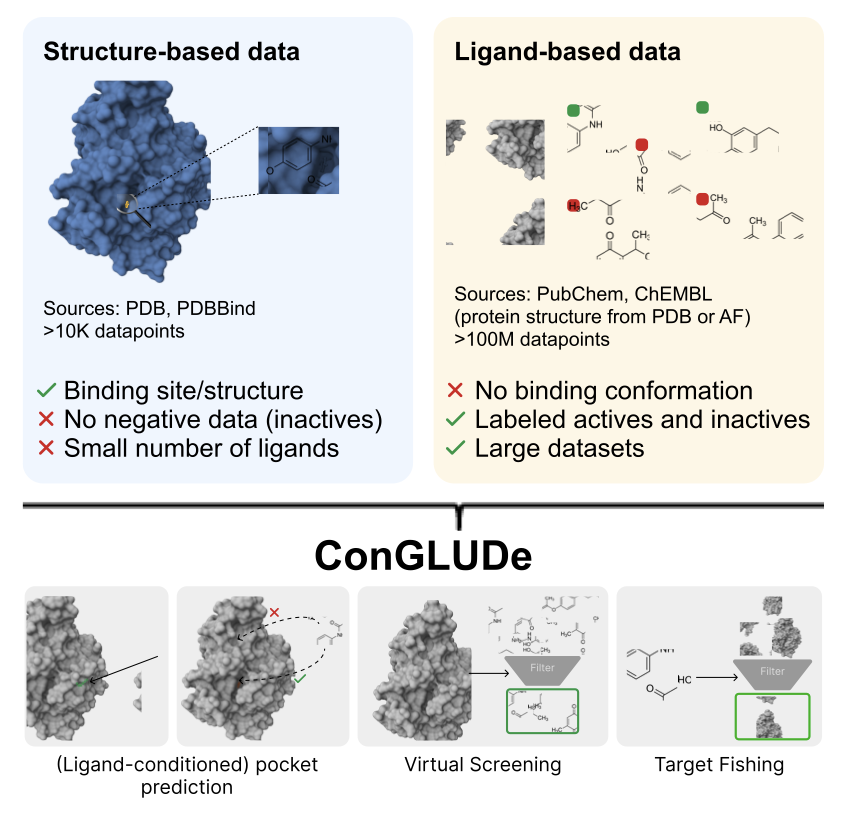
Contrastive Geometric Learning Unlocks Unified Structure and Ligand-Based Drug Design
1. A novel approach in computational drug design, ConGLUDe integrates structure-based and ligand-based training into a single model, overcoming traditional limitations of disjoint data sources. This unified method significantly enhances the efficiency and accuracy of drug discovery tasks.
2. ConGLUDe leverages a geometric protein encoder that predicts whole-protein representations and implicit binding site embeddings, removing the need for predefined pockets. This innovation allows for ligand-conditioned pocket prediction, virtual screening, and target fishing—all within a unified framework.
3. The model achieves state-of-the-art performance in zero-shot virtual screening without binding pocket information, outperforming existing methods on challenging target fishing tasks. It also demonstrates competitive results in ligand-conditioned pocket selection, highlighting its versatility.
4. ConGLUDe's training involves alternating between structure-based and ligand-based batches, utilizing a novel three-way InfoNCE loss function. This approach aligns protein, ligand, and binding site embeddings, enabling efficient large-scale screening.
5. The study evaluates ConGLUDe across diverse benchmarks, demonstrating superior performance in tasks like virtual screening and target fishing. The model's ability to generalize across datasets underscores its potential as a foundational tool for drug discovery.
6. Limitations include uncertainty in performance on proteins with predicted structures and the current lack of support for phenotypic assays. However, the potential for future extensions, such as integrating generative models for ligand design, suggests exciting prospects for this approach.
📜Paper: https://t.co/wwUPXBC71q
#ContrastiveLearning #DrugDesign #ComputationalBiology #AIinPharma #ProteinLigandInteractions
CryoDDM: CryoEM denoising diffusion model for heterogeneous conformational reconstruction
1. CryoDDM is a novel denoising diffusion model designed to enhance cryo-EM single-particle analysis by preserving high-frequency structural information while removing noise, significantly improving the accuracy of protein conformational heterogeneity classification and reconstruction.
2. The model introduces a two-phase diffusion process tailored for cryo-EM images, overcoming the limitations of Gaussian noise assumptions and reducing computational costs by optimizing diffusion steps.
3. CryoDDM outperforms existing methods by enabling high-resolution reconstruction of diverse proteins, including a proteasome, a membrane protein, and a spike protein, revealing previously undetected conformational states and motions.
4. The study demonstrates CryoDDM's ability to enhance downstream analysis, such as particle picking and 3D classification, by providing cleaner images without sacrificing structural details, thus advancing structural biology research.
5. CryoDDM's effectiveness is validated across multiple datasets, consistently showing superior performance in capturing dynamic protein behaviors and improving reconstruction quality, making it a valuable tool for cryo-EM studies.
📜Paper: https://t.co/NYrKaLOXlF
#CryoEM #Denoising #DiffusionModel #ProteinStructure #CryoDDM #StructuralBiology
NEURIPS! I just arrived! Check out our group's papers and find me + my students to talk science!
- CryoBoltz ❄️⚡️: Multiscale guidance of protein structure prediction with heterogeneous cryo-EM data
@rishwanth_raghu https://t.co/WDnJQgQZqL
- ChefNMR 🧑🍳⚛️: Atomic Diffusion Models for Small Molecule Structure Elucidation from NMR Spectra
@zyxiong123 https://t.co/pOPPdcqdR3
Workshop papers @workshopmlsb:
- CryoNOO❄️🚫: Separating signal from noise: a self-distillation approach for amortized heterogeneous cryo-EM reconstruction
@MinkyuJeon19791@jeffhygu
- CryoHype❄️🙌: Reconstructing a thousand cryo-EM structures with transformer-based hypernetworks
@jeffhygu@MinkyuJeon19791
Really excited to share these works. Separate tweet threads soon!