In a new work with @Josephmrich and Conrad Oakes we tackle the problem of how to best organize alluvial plots. We formalize two optimization problems and develop a solution for them based on the neighbornet algorithm, implemented in the program wompwomp: https://t.co/njQRkjYHNh
The authors report that all seven tested deep learning models failed to outperform simple baselines in predicting transcriptome changes after single or double perturbations.
I will duck for cover.
I’d love to hear biologists weigh in on the fundamental question: can you predict the impact of cell perturbations better by studying the natural variation in a population of healthy cells (more data), or by studying cells that have been perturbed genetically or chemically?
🤝Excited to announce @ProjectBiomni × @AnthropicAI!
AI agents are set to transform how biologists do everyday research. Thanks to this partnership, the platform is now free for scientists worldwide: https://t.co/9T2bOft1Nj
Learn more: https://t.co/Wh9SuToMm4
One thing that really bothers me with the new "virtual cell" terminology is that is currently largely focused on a very narrow definition of models that can predict effects of trans perturbations (gene dosage, drugs etc) on gene expression. 1/
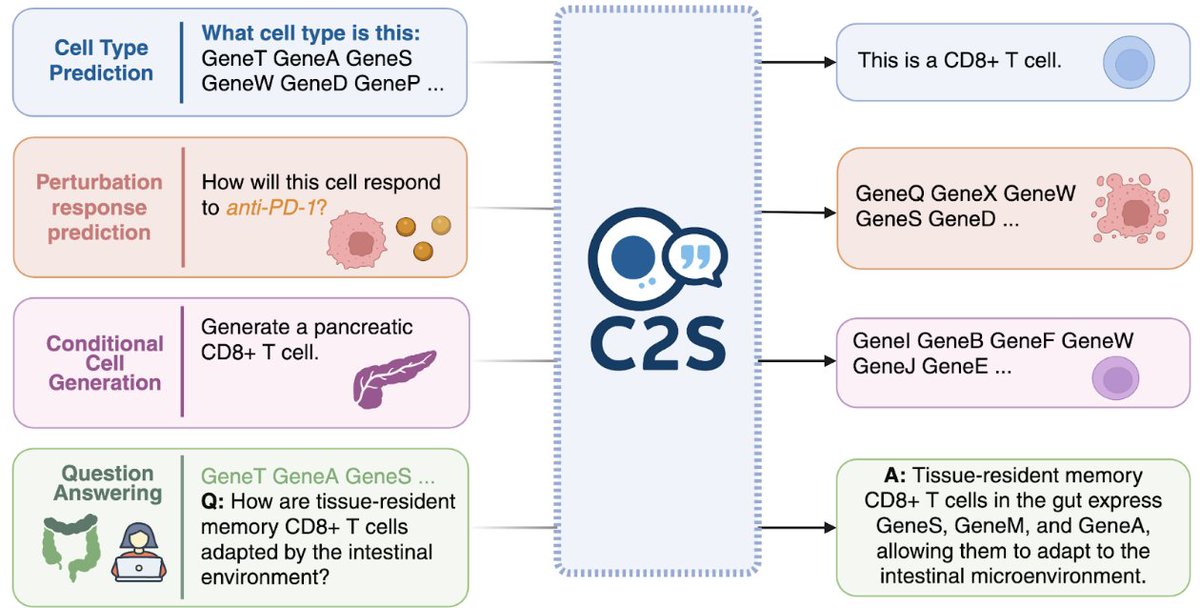
What if LLMs could “read” & “write” biology? 🤔
Introducing C2S‑Scale—a @Yale + @GoogleAI@GoogleDeepMind collab: we scaled LLMs (up to 27 B!) to analyze & generate single‑cell insights by turning transcriptomes into text 🧬➡️📝
🔗 Blog: https://t.co/3GbnXbKVmb
🔗 Preprint: https://t.co/beO8Z9CESc
#SingleCell #AI #LLM
🚨 Inflammation Atlas – Version 2 is here! 🚨
MAJOR update of our INFLAMMATION ATLAS, including >6.5 million single cells from 1,047 patients across 19 diseases.
Inflammation touches nearly every aspect of human health—from infections to cancer to chronic immune-mediated conditions. Yet, a universal, holistic understanding of inflammation has remained out of reach. With this release, we move one step closer.
Using single-cell genomics and machine learning, we chart the full spectrum of immune cell activation in peripheral blood. The data, generated from a simple blood draw, opens the door for universal, non-invasive diagnostics that can identify and classify inflammatory states across diseases.
With this study we lay the groundwork for #PrecisionMedicine tools that can transform how we #diagnose and #treat inflammation-driven diseases. Let’s harness our immune system's signals to build a new era of #diagnostics.
🔬 Congratulations to all our partners @humancellatlas@cnag_eu@DoctisEU.
https://t.co/qrhCpVaZsf
First paper from my PhD is online.
We noticed that copy number variation biases the differential analysis in the epigenomics data and developed a pipeline to address it.. we revealed new sets of chromatin regions relevant to Down Syndrome pathogenesis.
https://t.co/0o4WtTjbXq
Gene regulation involves thousands of proteins that bind DNA, yet comprehensively mapping these is challenging. Our paper in @NatureGenet describes ChIP-DIP, a method for genome-wide mapping of hundreds of DNA-protein interactions in a single experiment.
https://t.co/0aINMj2MTu
The informal poll results are in, & as I feared, most people are running ANOVAs in #R with functions that can give highly misleading (or flat out wrong) results. Follow along to see why using anova() and aov() is usually inadvisable & why you should be using car::Anova() 🧵
I am super happy my first first-author paper is out (https://t.co/ggIKpU09ob)! There’s even a thoughtful “news and views” along with it (https://t.co/0h9ZHV4gcb).
Here is my summary of our work in 20 tweets:
@tangming2005 CNV is worth noting however often overlooked in differential analyses! Thank you very much for tweeting my preprint! P.s. I've been following you to learn bioinformatics!