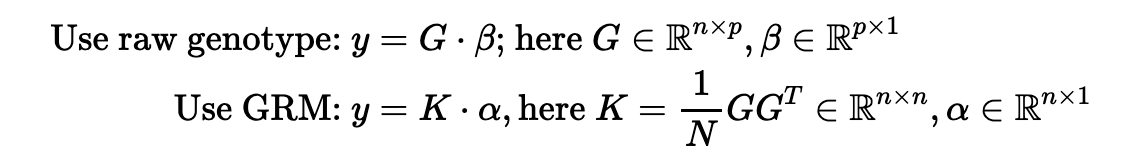@minouye271 Very nice work! Is it worth adding some convolution layers to account for LD? At least it should have some benefits of reducing # of parameters
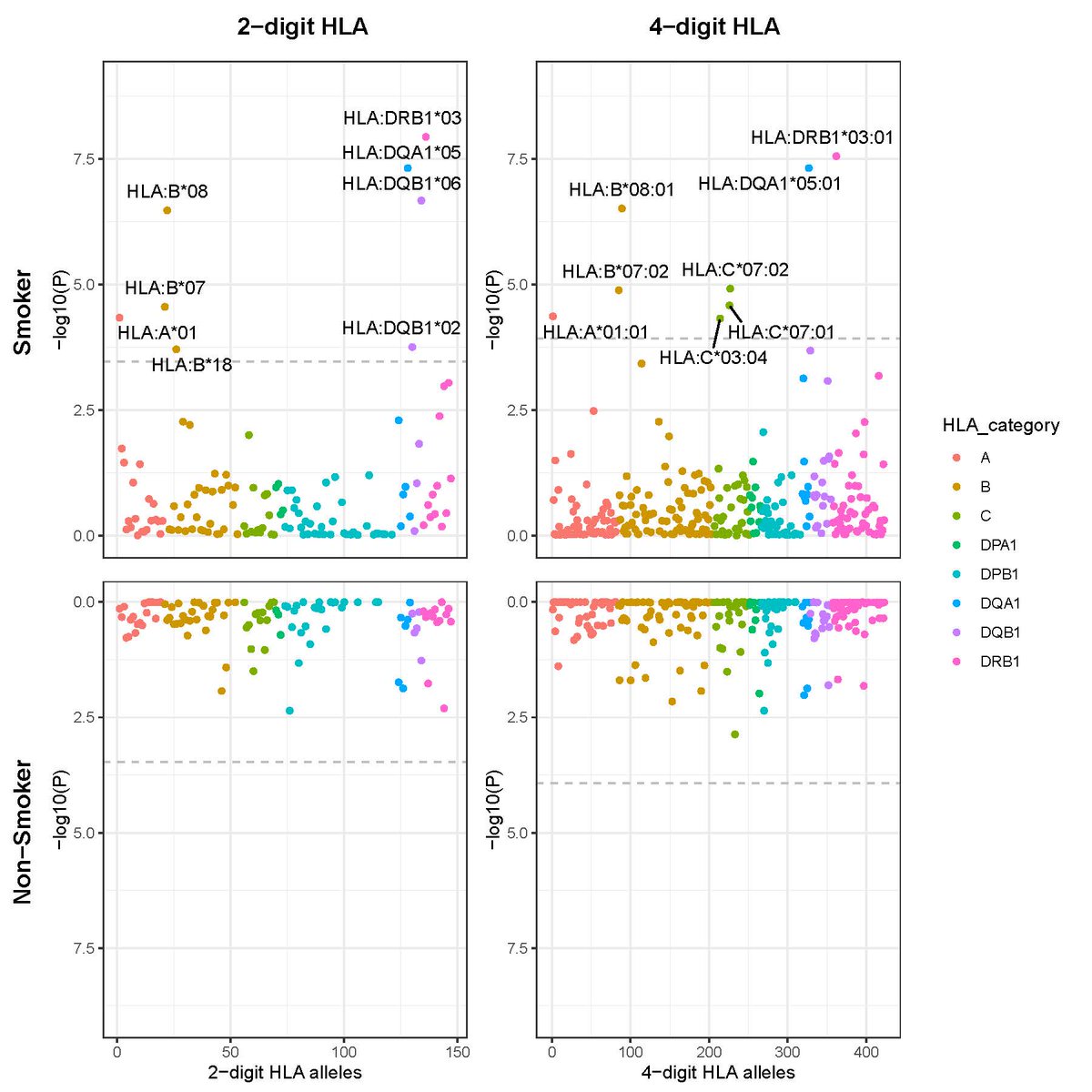
Happy to share new work with @anikethjreddy, where we aim to improve Enformer's gene expression predictions on personal genomes.
https://t.co/wqfLxgO8wM
@SashaGusevPosts I still find that "assuming each variant explains a tiny bit of variance" is quite clever. This way, the standard error for each marker is the same (condition on true effect size), which is 1/sqrt(N). By using iterated expectation, the expected chi2 can be worked out
@nmancuso_@KaiYuan1990 That’s great context. In 60s where genotyping is unavailable, this method can still be used for predicting traits. All we need is to keep track of the pedigree. Quite brilliant idea!
Is it possible to predict PRS using the genetic relationship matrix (GRM)? The GRM can be viewed as applying a linear kernel to the standardized genotype. Therefore, techniques such as kernel regression can be used. Moreover, one can modify GRM such that pairwise interactions
@KaiYuan1990 far as I can see, is the reduced dimensionality. Further, we can be creative in designing the kernel, even to incorporate interaction terms. This can be done by using the polynomial kernel, for example. The dimensionality will remain unchanged.
@KaiYuan1990 Just some raw thoughts:
PRS is modeling the joint effect size for each genetic marker, which is very high-dimensional. The kernel method says this linear model is identical to using a similarity matrix K (or GRM) as the predictor. This is called "duality". The benefit, as
LD is also computed in a block-by-block manner, which means if two variants are far away, their correlation is ignored. Would these ignored correlation, or LD, cumulatively play a role in determining the joint effect size?
A lot of stat-gen applications used a matched LD panel and summary statistics as a substitution for individual-level genotype and phenotype. e.g. the joint effect size \beta can be estimated with (X'X)^{-1} X' y, which equals D^{-1} \beta_{GWAS}. The D matrix, or the LD 1/n
panel, is often obtained from the 1000 Genome project, assuming the populations are similar enough. But how much variability would be induced by using an external LD matrix? I mean, we are estimating the entire LD matrix with only a few hundred samples...