Thrilled to start my independent career as an Asst. Prof. @ChemColumbia & an ARS @FlatironInst.
We'll develop new methods for modeling excited states, with an eye on practical photocatalysis. Other areas of quantum #compchem are also of interest, stay tuned for more!
The Rosen Group at Princeton University is accepting applications for a postdoctoral researcher interested in developing and applying machine learning interatomic potentials to model the dynamics of inorganic, solid-state materials. Apply at https://t.co/XQGr3dIoYW if interested!
GPU accelerated density fitting CASSCF from Ruiyan @mtzlab published at @JCIM_JCTC. Time for high-throughput, high-accuracy multireference simulations of ground and excited states!
Was a privilege to contribute.
#compchem
https://t.co/oZyIeowEoI
Yale Chemistry invites applications for tenure-track assistant professor positions in the following research areas: chemical biology, energy sciences, and physical or theoretical chemistry, to start July 1. Consider applying or sharing with your networks: https://t.co/YGTaN5Ttgm.
Please consider applying if it seems like a good fit or forward to someone who you think might be interested. I'm happy to chat about the institute, initiative, or my own research interests (electronic structure, photocatalysis, homogenous catalysis, etc.)
The Initiative for Computational Catalysis @FlatironInst invites applications at the postdoctoral level for Flatiron Research Fellowships. Fellows work alongside research scientists to develop new methods & software for studying electronic structure & dynamics of catalysis.
Please check out the initiative's webpage (https://t.co/3gbn7AHlYh) to learn more about us and our research, as well as the interfolio posting (https://t.co/C4kZ2y91cT) for further details. The deadline to apply is Dec 5 2025.
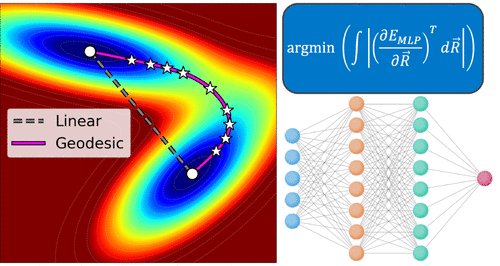
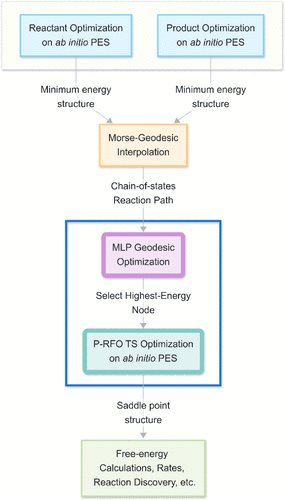
Identifying transition states often requires hundreds/thousands of ab initio calculations. We report a more efficient route by finding the shortest path (“geodesic”) between reactant and product on a machine-learned potential energy surface, without any ab initio calculations.
Geodesics on machine learned PES therefore offers a cheap & reliable route to finding TSs on ab initio PES. We invite the community to apply geodesic construction for finding transition states and would be curious to know what difficulties/limitations are encountered.
This has been a challenging problem that vexed me for a while. Congratulations to Richard (@mhg_group) and Leo (@FlatironInst) for leading this interesting work that opens up lots of fun applications (and challenges for even heavier elements).
https://t.co/GhN3SS2NRv
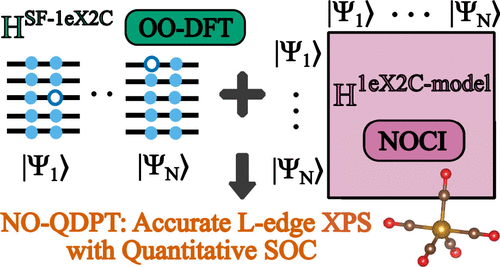
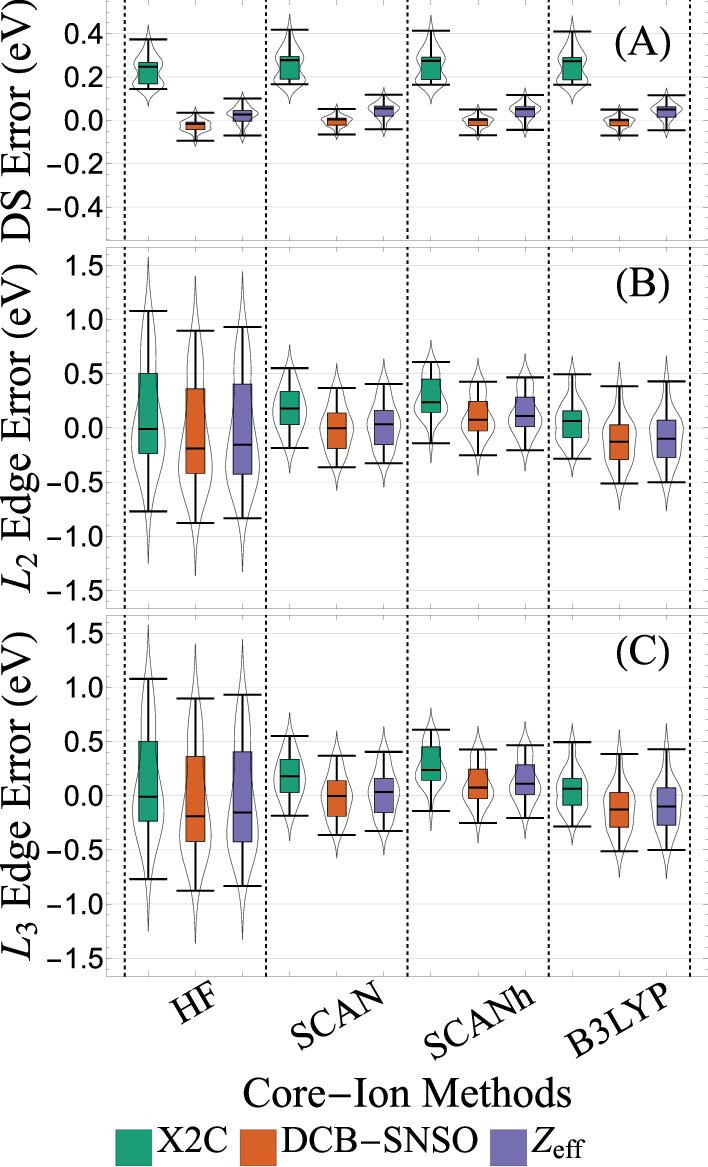
Predicting accurate XPS for shells with nonzero angular momentum is challenging because of spin-orbit coupling induced splitting.
We report a combination of OODFT and NOCI in @JCIM_JCTC (link below) that is accurate to ~0.2 eV vs expt. for L-edge of 3rd period elements.#compchem
@chenru_duan Thanks, Chenru! I saw your recent React-OT paper but still need to fully internalize all the math therein. Applications geodesics to optimal transport would be very interesting, please let me know if you discover anything along these lines.
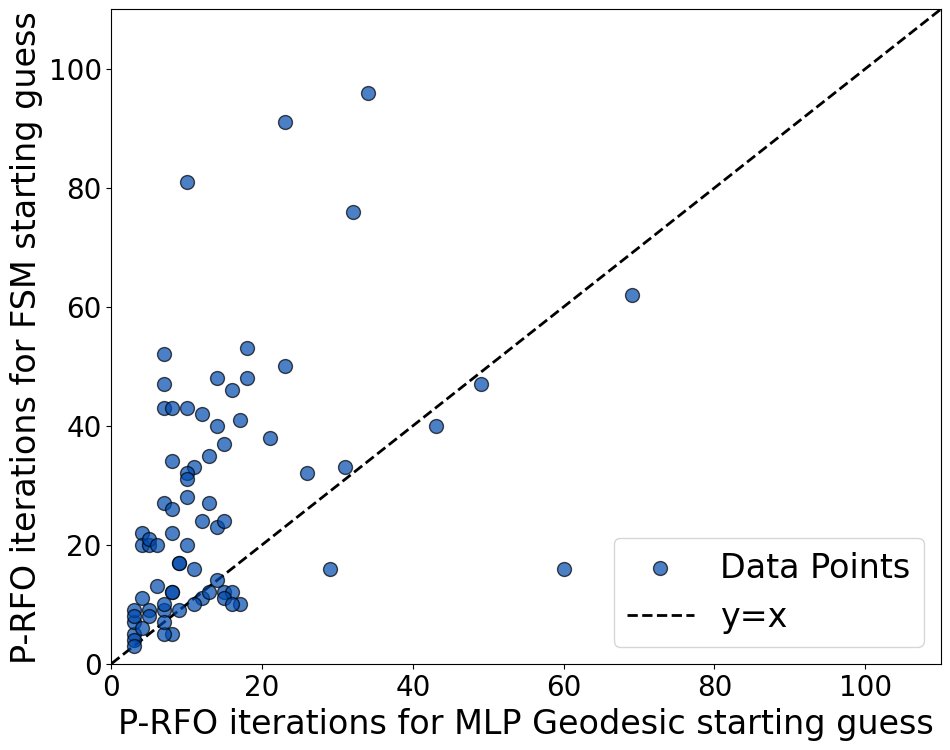
Locating transition states is often a #compchem challenge. We find that geodesics (shortest length paths) on ML PES can yield excellent initial geometries for transition state optimization on DFT PES (even compared to pure DFT frozen string method/FSM!). Preprint below!
Geodesic construction on MLPs is thus a cheap and effective way to initiate DFT transition state optimizations.
https://t.co/9Qh9p2GbAz is the associated preprint (with lots more context). This is my last Stanford project, together with @_jdep_, Martin Stöhr & @toddjmartinez.