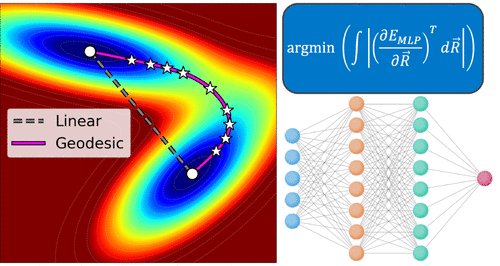
Identifying transition states often requires hundreds/thousands of ab initio calculations. We report a more efficient route by finding the shortest path (“geodesic”) between reactant and product on a machine-learned potential energy surface, without any ab initio calculations.
Excited to share @mtzlab 's venture into periodic quantum chemistry. All-electron hybrid DFT on unit cells with hundreds (or few thousands) of atoms in the timescale of hours, with quadratic scaling vs system size. GPUs+Gaussian orbitals ftw. #compchem
https://t.co/KC30Or3rXK
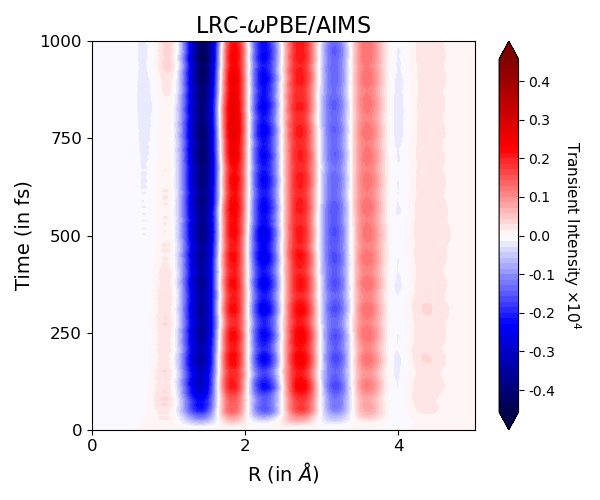
Check out our new collaborative study @JPhysChem A on probing coherent vibrational dynamics of CBr₄ with XUV spectroscopy of Br 3d electrons. Experiments by the Leone group @UCB_Chemistry and theory by our postdoc @fullCIalchemist
https://t.co/muSAFG8gFg
My research group at @USC is #hiring. We have multiple PhD and postdoc positions in Spring and Fall 2025. Interested candidates can email me with CV and research interests. Check our website for details: https://t.co/n77UGWVR2h. Please kindly RT 👍
#USC#PhDposition#Postdocs
Check out our new publication by our alumni @EliPieri90@arwalker_on_gpu and talented graduate student Mingning Zhu @nancyzmn !
Conical Intersection Accessibility Dictates Brightness in Red Fluorescent Proteins | Journal of the American Chemical Society https://t.co/JkNhRYp9Bp
Our contribution to the cyclobutanone photochemistry prediction challenge has been published @JChemPhys. Using EOM-CCSD & TDDFT with ab-initio multiple spawning, we estimate a few ps lifetime for the S2 state, leading to relatively slow photodissociation.
https://t.co/rAkYAK9puf
Delighted to share that I’ll be joining @USC as an Assistant Professor of Aerospace and Mechanical Engineering starting in January 2025. @USCViterbi@uscviterbi_ame
I’m indebted to my advisors, colleagues, and collaborators who have supported me to reach this career milestone.
Quantum Chemistry at massive scale! Our new open source systems, BigChem and ChemCloud, parallelize calculations across hundreds of nodes (or GPUs) simultaneously while achieving linear performance gains. Give them a try in your lab today🙌 @JChemPhys https://t.co/nlegWPzBEt
🎉 The MolSSI team extends our belated congratulations to Melisa Alkan, our former Software Fellow, on her 2023 ACS Division of Physical Chemistry Graduate Student Award in Theory! 🏆She is currently a postdoc @StanfordUChem @thecompchemist Well done! 👏😀
Check our new paper in @JChemPhys by our group alumni and members Roman Ellerbrock, @KGraceJohnson, @sseritan, and Harry Zhang. We released QuTree, a tree tensor network package available for public use. https://t.co/IJE7OuGHdY
Our new article in @JCIM_JCTC, led by Andreas Hillers-Bendtsen, presents a new strategy for achieving quartic computational scaling of coupled cluster ground-state energies, by using tensor hypercontraction (THC) in cluster perturbation theory.
https://t.co/Sm3CQIOQKe
Check out our contribution to the prediction challenge for the photochemistry of cyclobutanone. Using EOM-CCSD & TDDFT with ab-initio multiple spawning, we estimate a few ps lifetime for the S2 state, leading to relatively slow photodissociation.
https://t.co/oT452oOeQ5
It’s official! Alice Walker is receiving the @NSF CAREER Award for the proposal: Computational Design of Fluorescent Proteins with Multiscale Excited State QM/MM Methods. Congratulations @arwalker_on_gpu! #WSUchemistry#CompChem
Interested in controlling the excited-state reactivity/selectivity of chromophores via electronic tuning? Check out our (@ListNanna , @toddjmartinez) @CommsChem paper where we use QM to reveal how P-ring engineering can impact GFP photoisomerization yields
https://t.co/HYP9Jcub9w
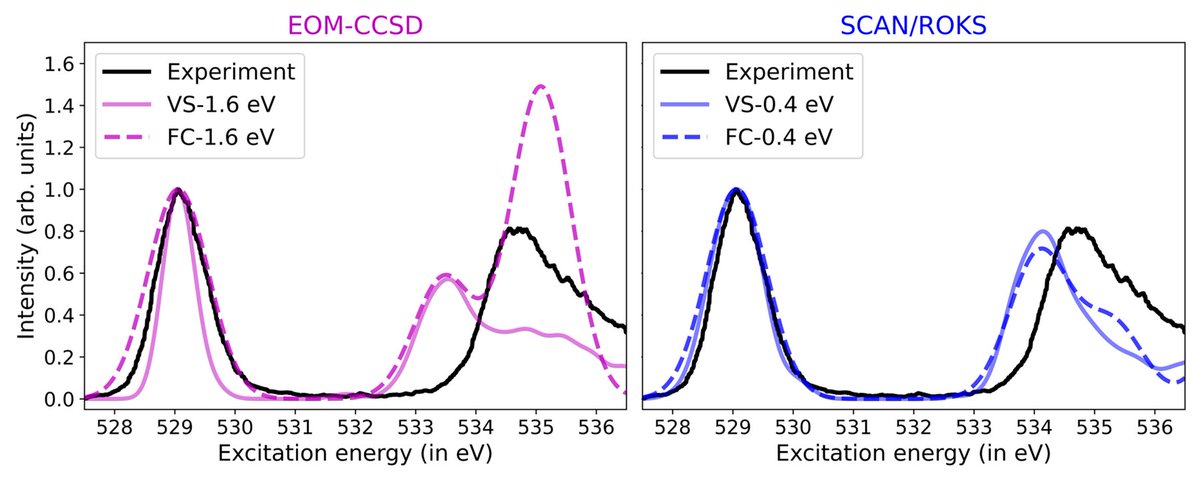
My first paper as a postdoc in @mtzlab just came out in @JCIM_JCTC. We wondered how well standard quantum chemistry can compute the XAS of ozone (prototypical biradicaloid) and found that orbital optimized DFT (i.e. ROKS) gives excellent results. #compchem
https://t.co/MPO4YZZ8jr
Interested in mechanochemistry? Our new collaboration paper with @YanXiaLab and @CraigGroup_Duke is published in @J_A_C_S. Great work led by Matias Horst @ladderchem !!!
Our paper on the nonadiabatic dynamics of polaritons are now available in @JPhysChem, https://t.co/jJGZJZrdIa. Led by @bhaskar_98, we applied ab initio multiple spawning (AIMS) to simulate the photochemistry of salicylideneaniline coupled to a cavity photon.
Hot off the press: More GFP engineering! Check out our latest article in @NatureComms to see how we (Yasmin Shamsudin , @arwalker_on_gpu , @toddjmartinez , Boxer Lab at Stanford) used QM/MM and MD simulations to engineer a more efficient split GFP!
https://t.co/lruvtgjEer