Our featured GCBR of the week is The Genome Aggregation Database (gnomAD) which aggregates and harmonises both exome and genome sequencing data from large-scale sequencing projects. Click the link to find out more https://t.co/fwrfmD1vFx
@NIH
I'm excited to announce that we have generated local ancestry informed allele frequencies for the inferred African/African American genetic ancestry group of gnomAD v4.0, live now on the browser!
https://t.co/KoOKdKRCcS
GeniE, the genetic prevalence estimator, is now available! https://t.co/QHtuIvLrGt
This tool allows users to estimate the genetic prevalence of autosomal recessive diseases using #gnomAD allele frequency data & classifications from #ClinVar
Blog post: https://t.co/aSpUglRKVj
gnomAD 4.1 is now live! This release fixes the AN issue in #gnomAD v4.0 & adds 2 new functionalities:
1) Joint AN across all called sites in exomes and genomes
2) A flag indicating when exomes and genomes frequencies are highly discordant
Learn more at https://t.co/ngnag9f1Bn
gnomAD 4.1 is now live! This release fixes the AN issue in #gnomAD v4.0 & adds 2 new functionalities:
1) Joint AN across all called sites in exomes and genomes
2) A flag indicating when exomes and genomes frequencies are highly discordant
Learn more at https://t.co/ngnag9f1Bn
Our paper describing a way to infer the phase of rare variant pairs using gnomAD v2 is out now in Nature Genetics.
We hope that the resource we generated will be useful when interpreting rare co-occurring variants in the context of recessive disease.
https://t.co/29Qflr6Yce
Thrilled to have our work on gnomAD out in print at Nature today. With 76K genomes, we can look beyond the coding genome and into the non-coding genome to find regions important for human disease https://t.co/6eVKy9rhwz
A special congratulations to @konradjk, @sc2643, @ksamocha and @MGoGuo for all their hard work on these publications. Read more about this work at https://t.co/X9Z7lpTXU2 (2/2)
The #gnomAD v3 papers are now published! This includes the non-coding constraint paper (https://t.co/akE9LGWymJ) & inferring compound heterozygosity paper (https://t.co/1Gw1sTrxWI). Congratulations to everyone who contributed to this valuable work! (1/2)
Hey all! We are running a survey to solicit broad-based feedback from users & *non-users* of Hail
Query. We'd like to hear from anyone who analyzes sequencing or genotype-chip datasets!
All questions optional, fully anonymous, & takes <=10min!
https://t.co/gMiV3snGBj
As part of v4, we are happy to announce the launch of the #gnomAD forum https://t.co/7Lkg4FHjFA. This will be a place for our users to help each other, discuss the data and ask questions. #ASHG23
To learn more about what is involved with QCing the gnomAD v4 dataset please attend Julia Goodrich’s #ASHG23 talk today (11/4) at 10:30am in ballroom B
QC of v4 required analyzing over 1 BILLION variants! More than 910 million variants passed our filters and are available on our browser https://t.co/YoRUWHD08N. More details on this variant dataset are available on our new stats page https://t.co/wzNXhlhiF1 (3/11)
Gene constraint is now available on #gnomAD v4! This is the first time we have had constraint data available on GRCh38. Katherine Chao will be covering this work during her talk at #ASHG23 tomorrow (11/4) at 11am in rm 202A.
To learn more about the impact of diversity on variant discovery and gene constraint please attend Katherine Chao’s #ASHG23 talk tomorrow (11/4) at 11am in rm 202A
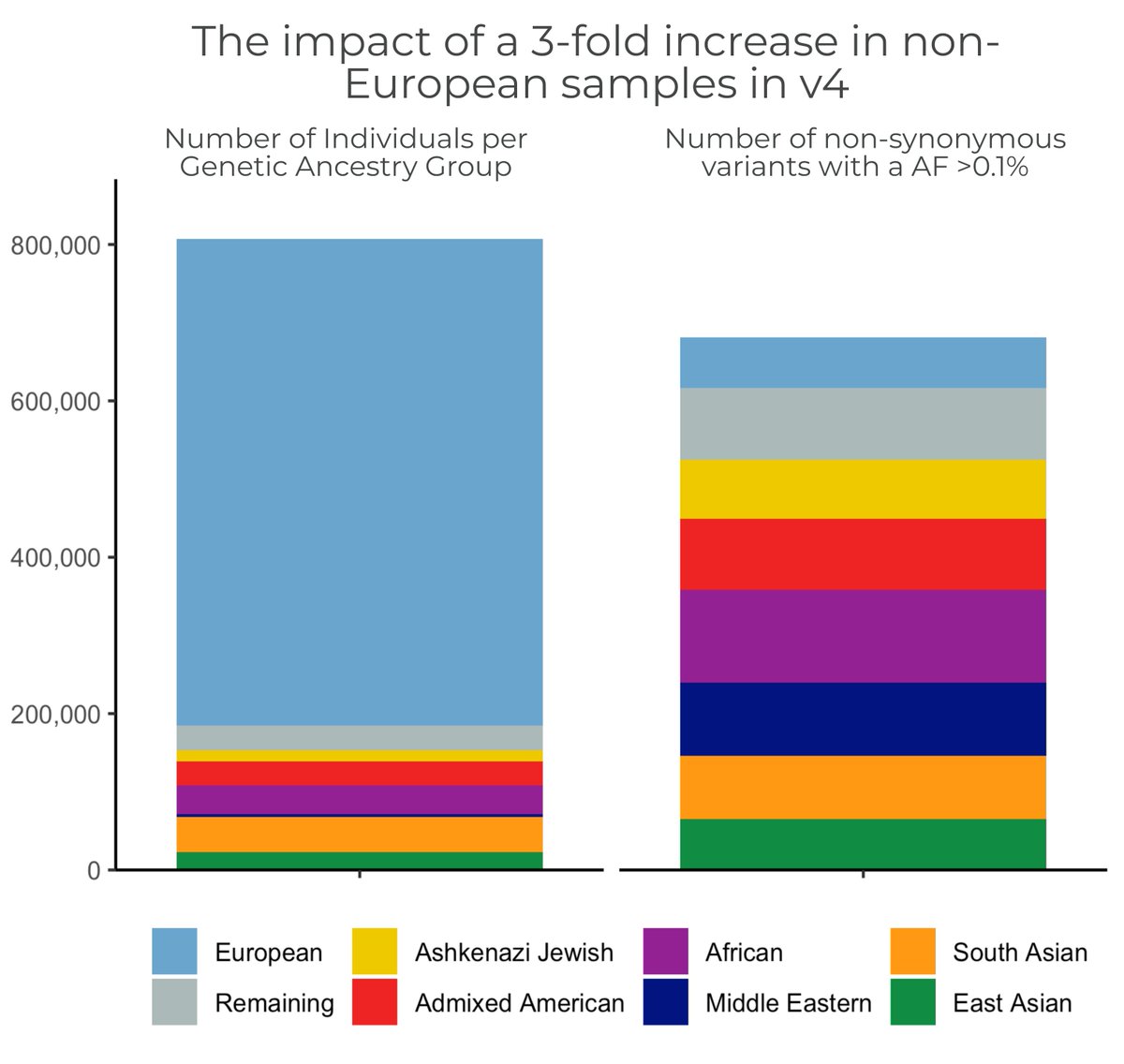
Our genetic ancestry blog https://t.co/lgqDr62oKc discusses our efforts to improve representation in #gnomAD, how we label groups and how the diversity in gnomAD is improving genomic filtration. (4/11)
To learn more about the technical details of how this data was generated read our blog posts https://t.co/WMh9WwPfWL and https://t.co/yCutZY8KHf. If you are attending #ASHG23 please attend Jack Fu’s talk today (11/2) at 1:45pm in rm 202A (2/2)
As part of #gnomAD v4, in collaboration with the @TalkowskiLab, we have released 1,199,117 genome SVs and 66,903 rare exome CNVs. These data represent the first gnomAD SV dataset released native to the GRCh38 reference genome. (1/2)
@hailgenetics@tpoterba @BroadGenomics Keep an eye on this account for the next few days to learn more about our methods including structural variation, genomic ancestry, and our QC process. In the meantime check out the v4 dataset at https://t.co/YoRUWHD08N (11/11)
The #gnomAD team is proud to announce the release of gnomAD v4! The v4 dataset includes 730,947 exomes & 76,215 genomes, which is ~5x larger than the v2 & v3 releases combined, & includes nearly 120K indivs of non-European genetic ancestry https://t.co/YKXIFlZwSi #ASHG23 (1/11)
@hailgenetics@tpoterba @BroadGenomics And of course, a special thank you to the 308 gnomAD data contributors and all the individuals who have enrolled in research. Without their willingness to share data and participate in research, gnomAD would not exist! https://t.co/oSaUWbWjia (10/11)