📄 Explore the latest from Bioinformatics Advances: "VueGen: Automating the generation of scientific reports"
Full article available: https://t.co/WWJYzaZoxW
Authors include: @sayalaruano, @Henrywebel
Passionate about genetics, metabolism, and neuroscience? Join us for a Postdoc to explore how obesity-associated genetic variants impact hypothalamic circuits using CRISPR-based Perturb-seq & in vivo metabolic phenotyping
https://t.co/XoDCdpK5bJ
@Metabolcenter#Obesity
@DemichevLab "We initiated this by conducting an entrapment experiment with a 1× larger shuffled human mimic protein database, analyzing single-cell samples in combination with 20-cell samples serving as a carrier proteome with default FDR parameter settings of 0.01 at protein level."
uv—you've probably heard of it, but are you using it for... pretty much everything?
Save time and speedup your workflows (in under 10 minutes) in my latest video.
00:00 - Introduction to uv
00:38 - Installing Python versions with uv
01:14 - Project initialization and setup
02:31 - Creating virtual environments
03:35 - Package management with uv
06:12 - Running Python projects with uv
Our article in Nature Communications! https://t.co/l1px8wjoU0 our DL tool Taxometer can improve taxonomy annotations for metagenome contigs using binning features and hierarchical loss, bridging the gap between taxonomy and binning
🎊 It has arrived 🎊, the 2nd edition of my "Deep Generative Modeling" book. It has 100 new pages, 3 new chapters (incl. #LLMs) and new sections. It covers all deep generative models that constitute the core of all #GenerativeAI techs!
Check it out:
💻https://t.co/ayRBQuPcyJ
What began as casual discussions and a small hobby project has now evolved into a full manuscript. We are excited to introduce you to "fastmixture", a much faster alternative to ADMIXTURE that doesn't compromise on accuracy in ancestry estimation.
https://t.co/HMV7Df6te6
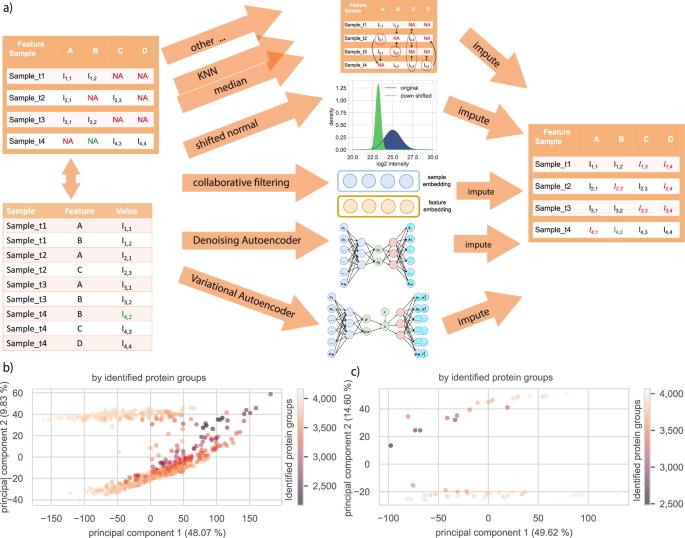
There, you can choose to set data aside for evaluation or not (with different shares of low to high intensities): Unsurprisingly during revisions missing-not-at-random (MNAR) and missing-(completly)-at-random (M(C)AR) came up as a topic.
The core functionality - three new models for imputation and the comparison workflow - is tested using integration and unit tests on a weekly basis, but please provide any questions or encountered errors in an issue: https://t.co/caFkEuCA7a (you will need a free GitHub account)
For easy integration of the three new PIMMS imputation models in custom notebooks or scripts, we provide a Scikit-Learn Transformer interface, i.e. a way to call fit and transform directly on your data. Try these in a tutorial on colab: https://t.co/QNypOVIpFV
Our work on introducing self-supervised models for imputation is out in @natureComms: https://t.co/YAk1VscBpr
We developed the models for MS-based proteomics data, but in principle they can be applied to any tabular data.
docs: https://t.co/pE2F1sz87r
https://t.co/HzmvZOVlzg
See an example comparison workflow including the downstream comparison in the paper here on a public alzheimer dataset (for protein groups): https://t.co/A6iDd6siG3
Imputation in mass spectrometry-based proteomics is a recurrent step of importance for downstream analysis. We offer an extensive comparison workflow of 27 established with three new scalable, fast and performant methods from deep learning for large and high-dimensional data.