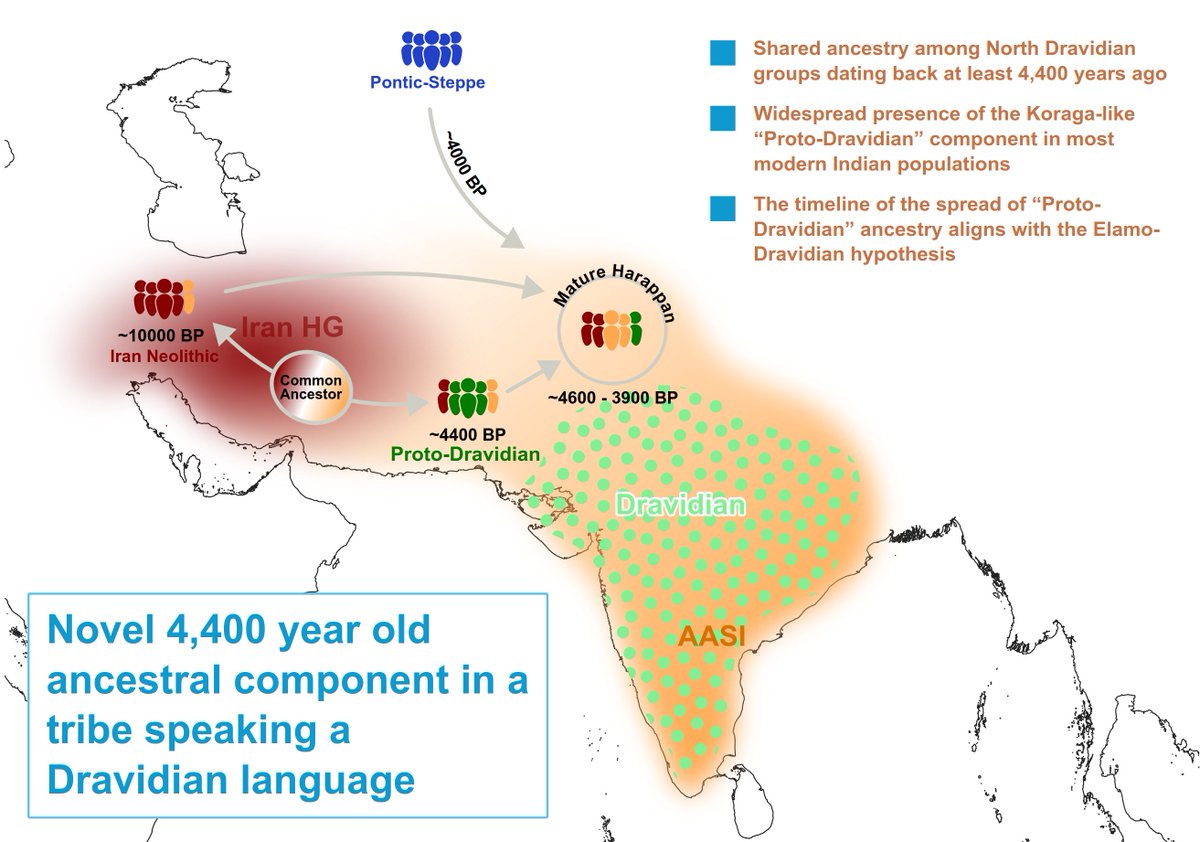
Our article describing the "Proto-Dravidian" ancestry is now peer-reviewed and published in @ejhg_journal There are several updates in the final version. You can read the full article using this link: https://t.co/q4MQuBFl0X #protodravidian#dravidian#ancientcivilizations#IVC
Wanted to follow up on this post and explain why archaic centromeres being found in modern people is so amazing. Also am I crazy or does one of the haplotypes look potentially super archaic in origin 👀
At a small but non zero fraction of your genome, you are closer related to and share a common ancestor with a Chimp / Bonobo before sharing a common ancestor with all other Humans. Pretty amazing!
Why associate haplogroups with religion? Looking deeper reveals the ethnic identity of local ancestry of the sample. Social, linguistic, and ethnic constructs have their limits, but they easily pre-date religious identities - the most fragile constructs of all!
The Old Lady spider cave skeletons in Ladakh have diverse maternal genetic origin.
• mtDNA diversity of the Cave site links to East Asia, Central Asia, and South Asia
• Cultural amalgamation of Buddhist (C4a1), Muslim (M52), and Central Asia (H2a1a)
https://t.co/SjFaHxALEO
Dryland farming communities inhabit the Deccan Plateau. One such farming family owns a vast amount of land and has traditionally lived as a joint family. According to DNA evidence, this pattern may have persisted all the way north, spanning the extent of the plains.
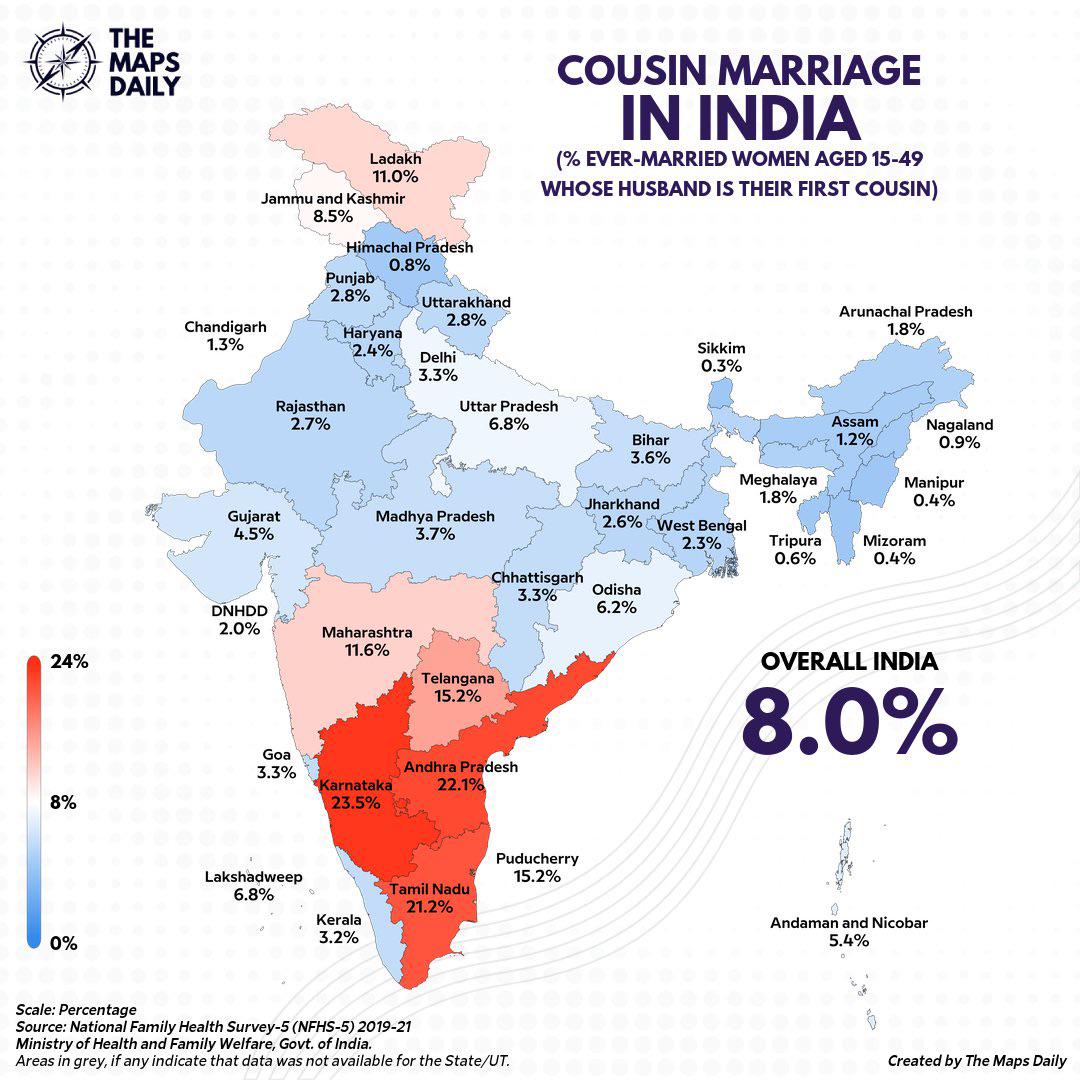
"Cousin marriage among Indian Hindus is relatively uncommon nationwide, estimated at roughly 9% overall, but it is highly regionalized. The practice is significantly higher among Hindus in South India—particularly in Tamil Nadu, Andhra Pradesh, and Karnataka..."
1/7
The last few days, we were busy running a workshop on Temporal Population Genetics ⌛🧬, hosted by the National Centre for Biological Sciences (@NCBS_Bangalore).
See the 🧵 for more details!
The shorter Denisovan haplotypes in South Asians are related to Den 25 (based on my analysis), while the longer ones are related to Den 3. It is highly likely that the two pulses observed in Southeast Asians occur because Den 3 and Den 25 shared a common ancestor. Awesome paper!
Unless we tease out ancestral components -specifically their formation and adaptation at highly localized levels -we simply aren't sampling correctly. Thus far, we have examined sequences from mixed groups; we must now look more closely at drifted populations
The idea that sequencing more genomes would lead to better medicine and better health was a good hypothesis in 2000. But 26 years later, evidence has quite convincingly disproven that hypothesis.
The answer to most common chronic illnesses that plague us isn't written in genes. Personalized medicine likely cannot come from sequences of nucleic acids. There is more to life's dynamic nature.
Why do we cling onto that hypothesis/dogma like it is truth.
New paper out today in @NatureComms from @gokcumen37392's group presents evidence for selection for higher number of Amylase copies in Indigenous Andeans. It looks quite interesting and I need to read it more deeply.
Read the study here:
https://t.co/jCXCxpbLj5
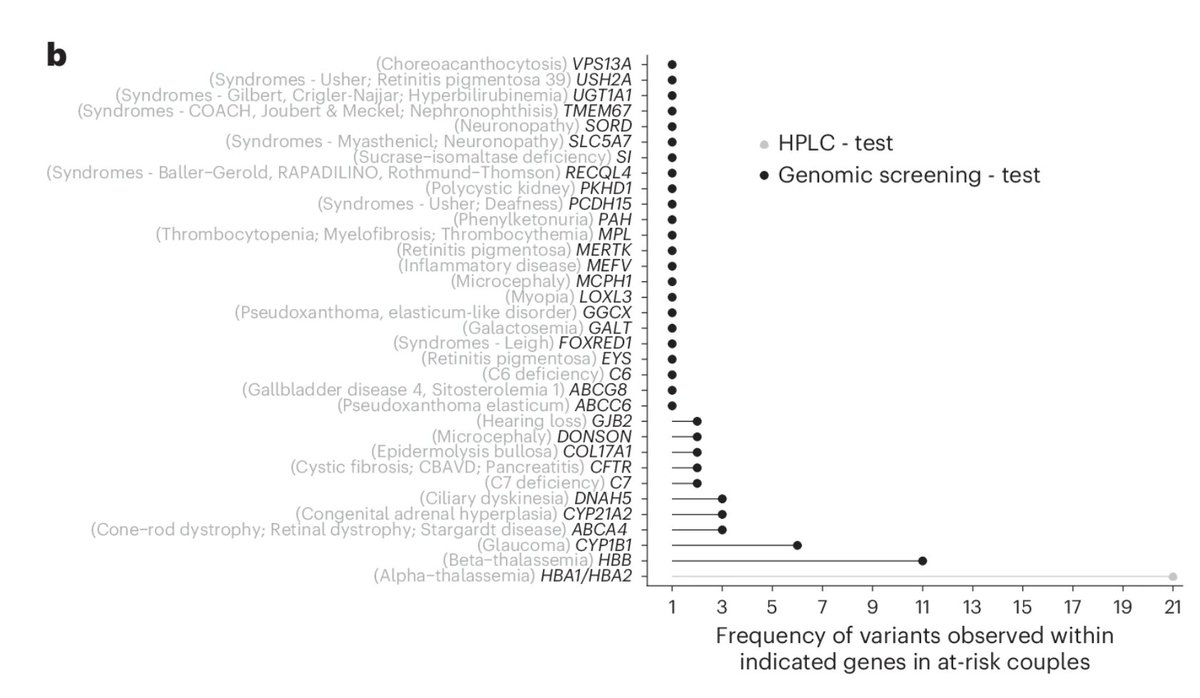
Interesting that the most common recessive disorder (alpha-thalessemia) in the UAE can't be reliably screened via standard short read sequencing.
From "Citywide premarital genomic screening in a Middle Eastern population"
https://t.co/8gdByd2VIc
The loss of talent and ideas due to the "Regular" employment requirement is a huge blow to Indian R&D. Why not allow the "temporary" to apply for project grants, with a clause ensuring the host institution maintains their employment for the duration of the project? @ANRFIndia
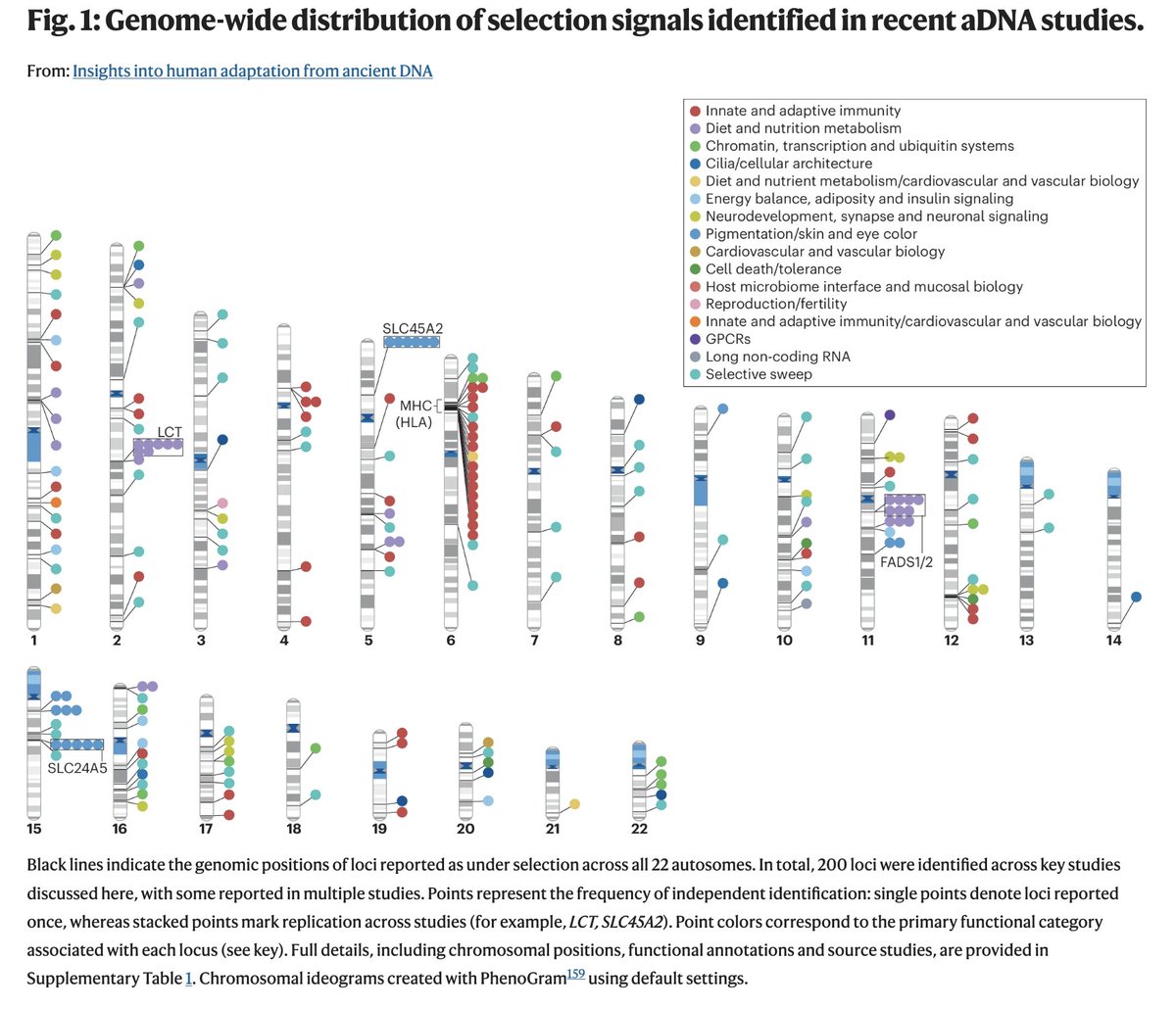
A new review article looking at what ancient DNA tells us about selection from @christiandhuber's group was published today in @NatureGenet
Read it here: https://t.co/PMElBiWJel
"I want you to explain f2, f3, f4 statistics using an intuitive visualization in time-frequency space."
An altogether fine version.
When I made similar visualizations by hand I made the lines squigglier to show stochasticity of allele frequency changes...
1/2
New preprint led by Hrushikesh Loya, Leo Speidel, and I where we introduce GhostBuster! https://t.co/MgfPEVeHQJ
Our method uses genealogies to find "ghost" ancestries hidden within DNA. We find both modern humans, Neanderthals formed as mixtures of two ancient hominin groups
It will be interesting to see which communities shifted from matrilocality to patrilocality (especially in India). Even in tribes that adopted patrilocality due to societal pressure, women often remain the primary decision-makers, particularly over ancestral property #societies
Global distribution of present-day societies that report patrilocal (green circles), matrilocal (purple triangles), ambilocal, avuncular, and neolocal (white, yellow, and orange squares, respectively) post-marital residence patterns.
Read this commentary by @doctorveera on the recent paper by @aliakbari23 and their team. Additionally, I would like to mention the role of the so-called 'conserved regions' in all of this. Archaic haplotypes have been conserved for reasons we have yet to discover. #selection
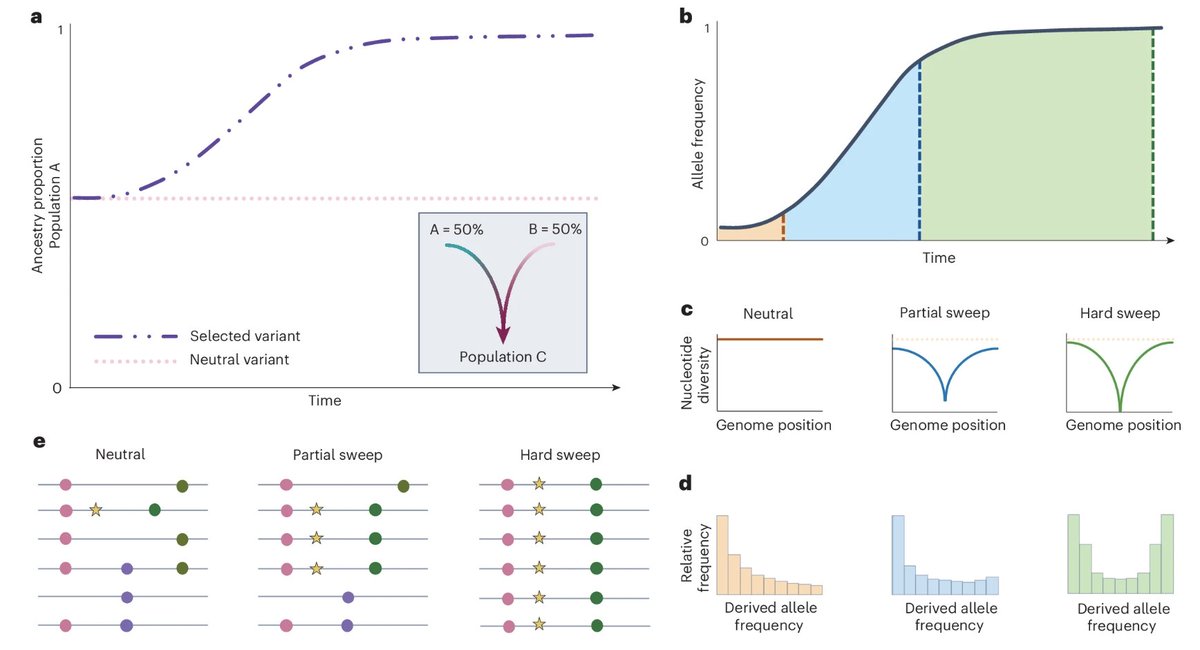
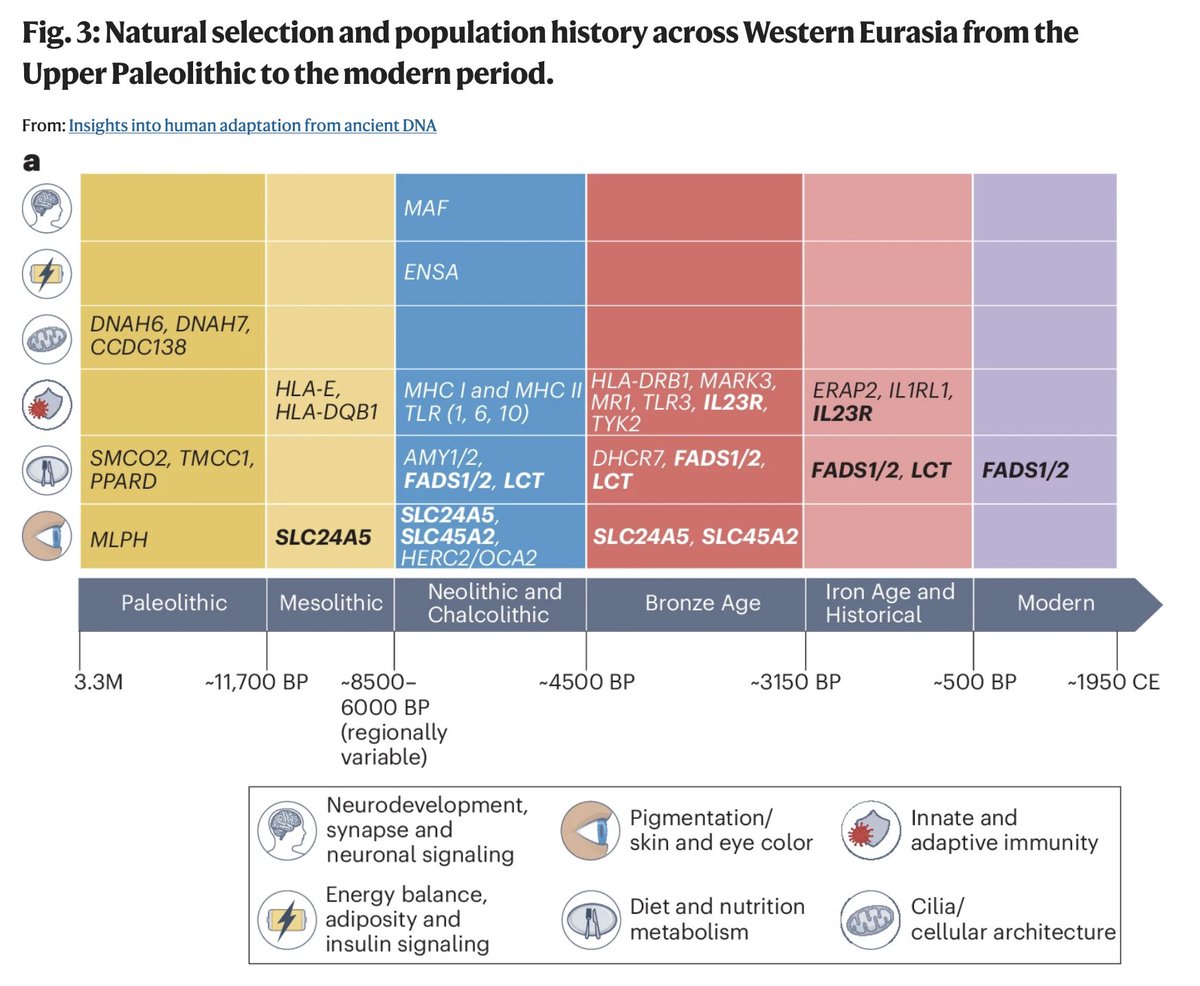
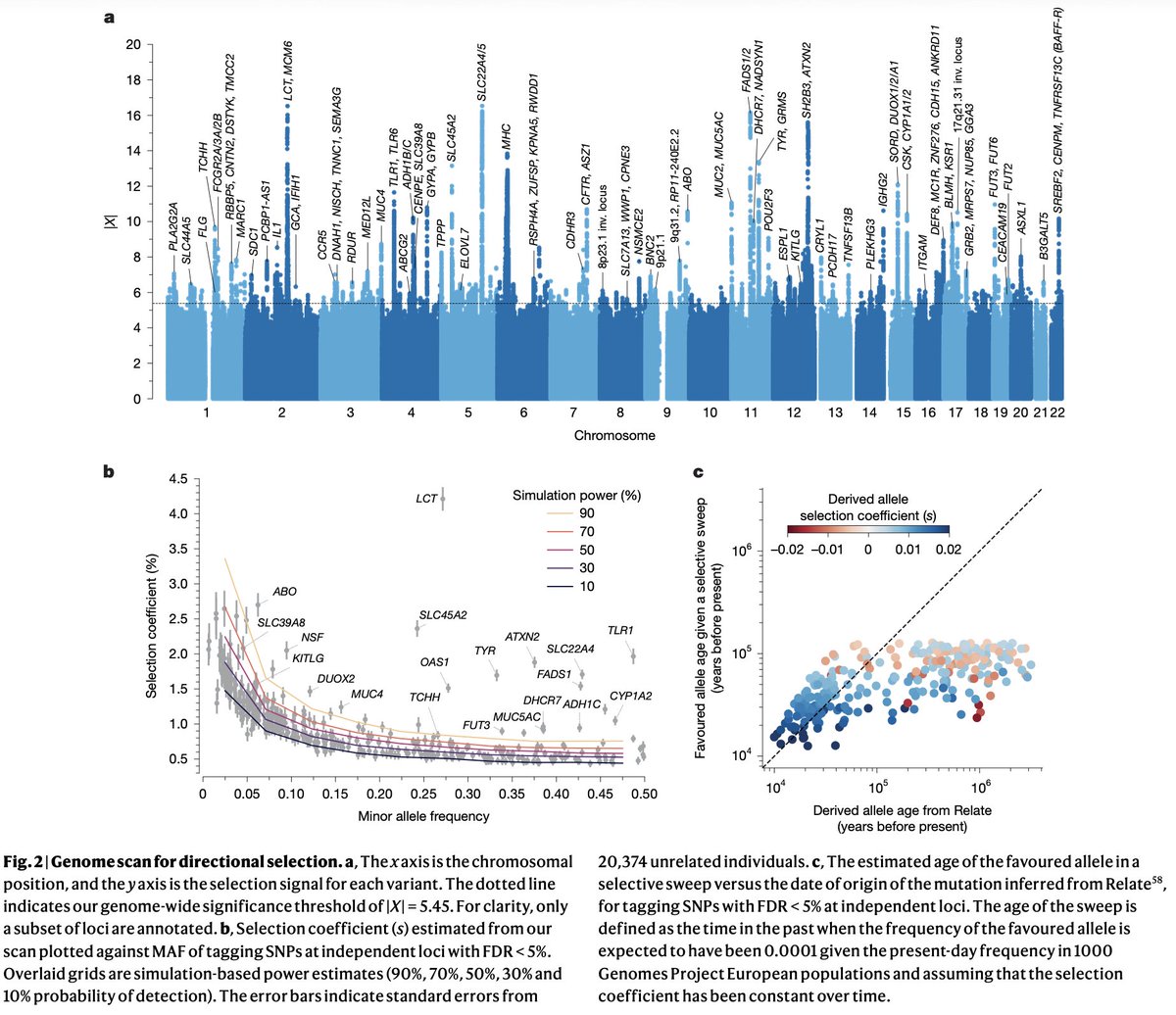
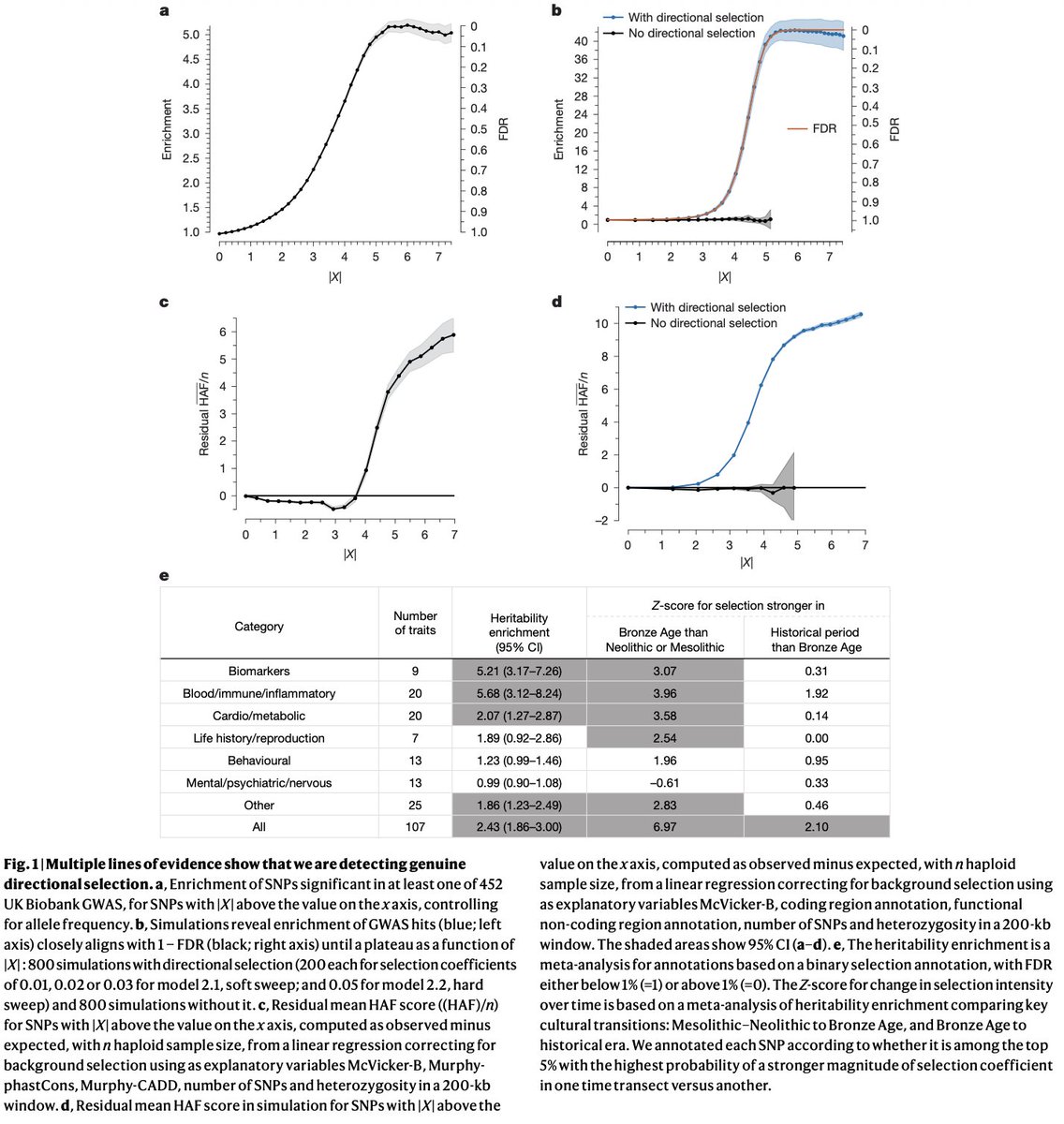
A new paper in @Nature from David Reich, @aliakbari23 and colleagues breaks the conventional understanding of recent human evolution. The field believed that strong selection in the recent past (~10,000 years) was rare, with few exceptions like the lactase persistence locus. In this paper, the authors challenge that belief, showing that we weren't looking at the problem right.
Previous studies that looked for evidence of selection using ancient DNA addressed the problem cross-sectionally, asking if allele frequencies differed across populations more than what one would expect based on genetic drift and migration. Most arrived at the conclusion that population structure primarily explained the observed differences. Here, the authors addressed the problem longitudinally, accounting for when ancient individuals lived by explicitly modeling time as a variable in the analysis. It turns out doing it this way dramatically increases power, increasing the number of genome-wide significant selection signals by 20-fold!
Looking at why accounting for the time variable led to such dramatic changes in results, the authors find that previous studies missed so much because selection often happened not on new variants leading to dramatic sweeps (the conventional model: new variant -> selection -> increase in frequency) but on already existing variants driven by transient environmental pressures. Many of these variants underwent reversals, selected up when a pressure existed, then purged when it disappeared or the trade-off cost became dominant. A great example is the TYK2 variant, where an allele boosting immunity was selected for thousands of years because it protected against TB, then got purged as TB endemicity declined and the autoimmune cost took over.
The scale of what they found is striking: hundreds of loci showing strong selection in the past 10,000 years with a median selection coefficient of ~0.86%. This number is pretty big in evolutionary terms, meaning allele frequencies have been shifting by ~1% per generation in a consistent direction. Previous selection scans found a maximum of 20 loci, and this one finds hundreds. That isn't an incremental change. It fundamentally reframes our understanding of how common strong selection has been in recent human history.
Some of the most striking findings come from polygenic selection, where hundreds of small-effect alleles were pushed in the same direction simultaneously. Polygenic scores based on large-scale GWAS of today predict recent negative selection for traits like body fat, waist circumference and schizophrenia, and positive selection for others like cognitive traits. One important caveat is that GWAS phenotypes are measured in industrialized societies today, and how well they capture what was actually being selected in ancient environments is debatable.
For me personally, these findings have direct implications for drug discovery. When using human genetics to find drug targets, we often fixate on the benefit and risk profiles of variants visible today. But we need to be aware that a variant's benefit:harm ratio might be environmentally contingent, and could reverse when the wrong environment manifests. An evolutionary understanding of a variant's association with traits is therefore essential.
The same logic applies, perhaps even more urgently, to embryo selection. Selecting embryos based on polygenic traits is humans making permanent, heritable decisions for their offspring with a narrow view of today's environment. The ancient DNA record now shows that cost-benefit landscapes flip over time. So, an embryo carrying man-made selections is carrying those changes into an unpredictable future environment.
The broader takeaway is that human evolution didn't freeze in the last 10,000 years. We just lacked the tools and datasets to see its movement. The current findings are based on European populations. I am curious to see these analyses extended to other populations too, like South Asian, East Asian and African populations, which might be holding more surprises to blow our minds.
Akbari et al. Nature 2026
https://t.co/3WWjpTiVgA
Its finally out! Over 8 years of work by @aliakbari23 shows that selection was pervasive in the last 10,000 years, including for quantitative traits. This was directional selection. Simulations show background selection, etc can not produce the observed patterns.
Take Pop 21 (DR) and Pop 25 (IE). At K=8, a shared "light pink" component (persistent through K=13) bridges these groups despite different language families. Their short/numerous ROH and private X-variants prove this isn't recent drift, but an ancient, stable substrate. 8/n
GIP results are out. The beauty of human diversity in India hasn't faded a bit. Out of the 5000-odd communities, the largest genetic dataset for India includes only about 80 pops. This means these results are just 1.6% of the whole story 1/n #india
https://t.co/OkweoZGf6A
The lowest CV error begins to plateau from K=8, not K=5. For exploring true fine-scale structure in GIP, the ADMIXTURE plot must be read from K=6 onwards. This is precisely when we see internal structure within the paper’s defined biogeographic regions. 7/n