Next #BSGM2024 plenary session on gene therapies. First talk @RJYanezMunoz president @_BSGCT, providing an overview of clinical application of gene therapies in rare and common diseases ➡️ great @ISCTglobal resource on approved gene therapy products: https://t.co/L2hqkPf0of
Long non-coding RNAs comprise a large part of the genome, yet loss of one copy (haploinsufficiency) of a lncRNA is not known to cause a human disease. Read our preprint to learn how deletions of the lncRNA CHASERR cause a severe neurodevelopmental disorder.https://t.co/JgjDvIcwer
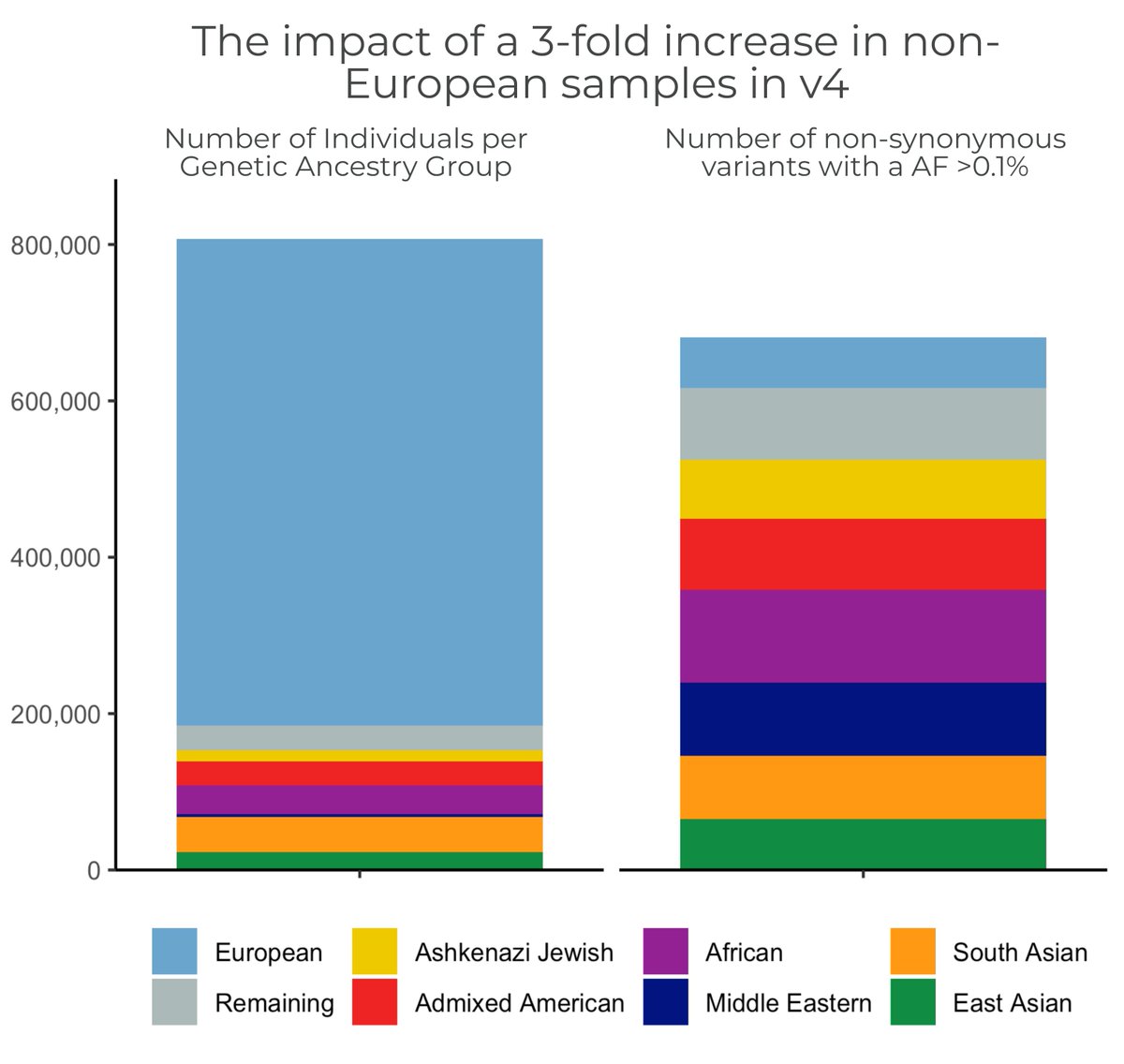
The released set has ~800k samples, but we started with a 955,000 sample call set! *Staggeringly* large!
We developed the Scalable Variant Call Representation (SVCR) and implemented it as the Hail VDS to enable this nearly 1M sample callset.
1/3
The #gnomAD team is proud to announce the release of gnomAD v4! The v4 dataset includes 730,947 exomes & 76,215 genomes, which is ~5x larger than the v2 & v3 releases combined, & includes nearly 120K indivs of non-European genetic ancestry https://t.co/YKXIFlZwSi #ASHG23 (1/11)
Finally, we show how to leverage this pQTL catalogue to prioritize missense variants in phenome-wide association studies. Combining PTVs with missense cis-pQTLs associated with decreased protein abundance increased the power to detect gene-disease associations
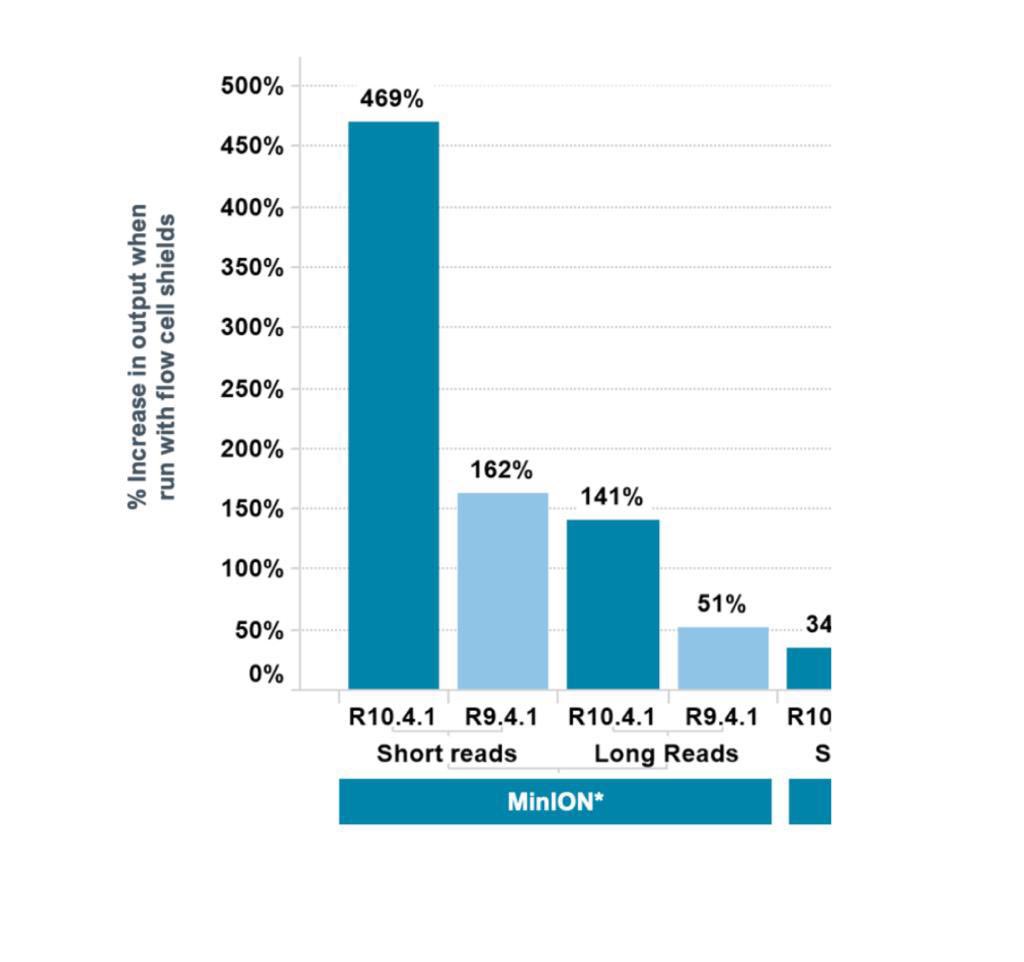
So @nanopore discovered something a few days ago - and it has to do with light. And they posted this image (sorry, too good not to share off the community). Yes, that’s 469% increased output. So, we decided to give it a shot and …
"Nearly 1 in 4 CSSVs [canonical splice site variants] may not cause LoF [loss of function] based on analysis of RNA-seq data" (N=121)
Oh et al. medRxiv
https://t.co/4DplJU1y7Z
- Are tandem repeat (TR) genotyping tools accurate beyond well-studied loci?
- How often do they over-/underestimate expansions?
- What % of variant loci are missing from existing genome-wide TR catalogs?
To answer these questions, we created a genome-wide TR truth set: [1/N]
New blog post!
https://t.co/TAiNKMVxGl
I deal with a lot of tab-delimited data in bioinformatics, so I recently spent some time putting together Bash functions for viewing them on the command line. Might be useful to others!
(1/5)
Determining the phase of two variants is a key part of recessive diagnoses, but is challenging to do with short-read sequencing data and often requires parental data.
We used gnomAD to aid in phasing, and explored compound heterozygosity in these data 1/
https://t.co/zdZPAOIjmm
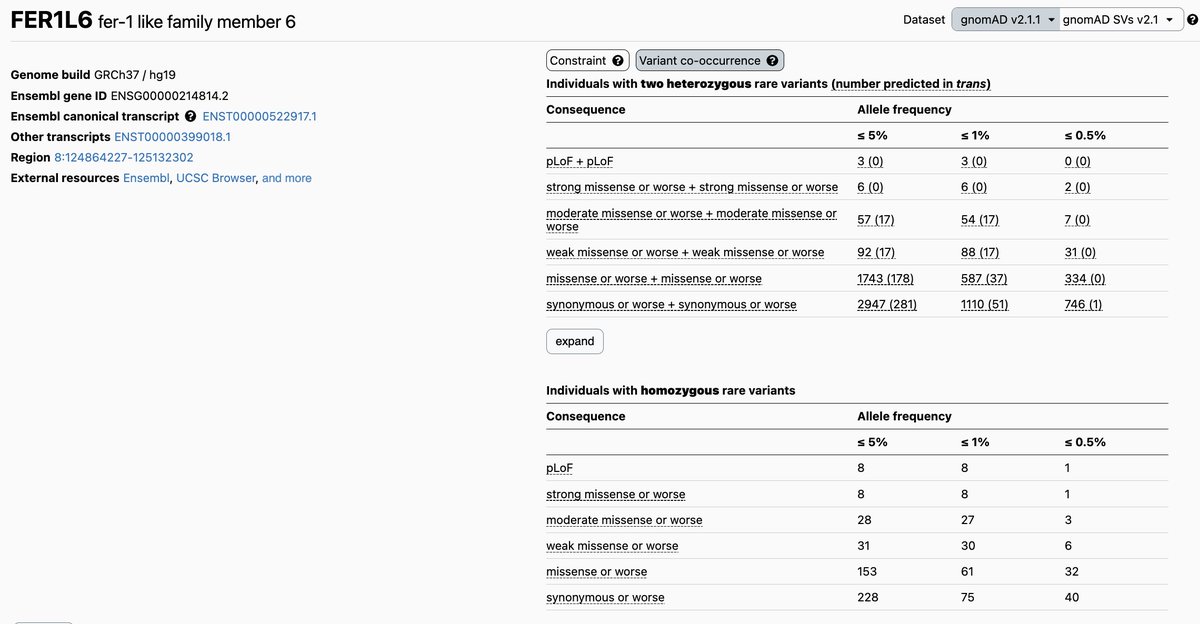
The #gnomAD browser now includes variant co-occurrence counts by gene. We hope this will help with interpretation of co-occurring variants in the context of recessive conditions. Read more about this feature on our blog post https://t.co/2crfyFxYde #ACMGMtg23
In a packed room #ACMGMtg23 with @HeidiRehm Les Biesecker and Steve Harrison presenting the next version of variant classification work at @TheACMG@ClinGenResource AMP and CAP. Version 4 will be points based. All slides available at https://t.co/tnIztMheej
Huge congratulations to Prof Matt Hurles (@mehurles ) who has been appointed as the new Director of @sangerinstitute! 🎊
Currently Head of the Human Genetics Programme, Matt will formally take up the Directorship later this year.
Find out more⤵️
https://t.co/qroO8pEUoi
📢STP Equivalence funding available for 200 healthcare scientists 📢
Along with the @ahcsuk we are delighted to announce an exciting initiative that aims to increase the number of statutory registered healthcare scientists.
ℹ️
https://t.co/nqAceYyyIb