@mihirbafna14 and I are excited to introduce Promera, a co-folding and design model with
• best-in-class binder filtering
• nanobody design with in-silico success rates matching hallucination
• case studies on hantavirus epitope targeting and GPCR agonism (1/8)
Today we’re launching Claude Fable 5, a Mythos-class model made safe for general use. Fable 5 is far better than any model we’ve ever released on long-running tasks. Disentangling bio capabilities from risks is hard, so Fable 5 ships with safeguards that block responses in biology. These queries will receive responses from Claude Opus 4.8.
We're investing in lab-grounded red-teaming so our biosafety calibration reflects actual threat models. That’s how we'll drive down false positive rates without lowering the bar on risks.
We also plan to open a trusted access program soon for select life science organizations to access Mythos-class models for biology and chemistry use.
Personally, Fable 5 has completely changed the way I work. Claude’s vision is now the best in the industry. I routinely let Fable cook for hours at a time on complex tasks without checking in, and it makes sensible choices. I’m excited to see what users do with this model. As always, my advice is to try a bunch of hard tasks that have never been possible before and see what you find.
This is also the first model release that my team has had a small part in, just two months after joining @AnthropicAI. I’m insanely proud of our team and grateful to everyone at Anthropic who has jumped in to collaborating with us, especially to folks who are brand new to the wild world of biology and drug development. More soon.
mythos will be bad ON PURPOSE on ai "frontier llm research" tasks, this is very very sad for the research community
also the fact that this is un purpose not visible to the user is crazy
Our preprint is out! We present the first generative pipeline for the de novo design of asymmetric β-barrel nanopores, enabling tunable pore geometries and hydrophobic thicknesses.
https://t.co/a4t0XO0GGZ
@ria_sonigra@sagardipm@lyjjj12138@FangleiXue
We ran Pearl on OpenBind, a benchmark created to challenge co-folding and assess the field. It surpassed all other models — zero-shot.
Our Pearl system exceeded the other models on every metric, particularly on the most relevant measures that correspond to performance on real-world drug programs.
More detail for fellow nerds👇
https://t.co/2jqJSBFi9p
1/ Zero-shot means zero added help.
Pearl got a protein sequence, a ligand SMILES, and an unbound template. Nothing else. (We also tested it providing a few binding-site residues, but zero-shot performance was already outstanding.
→ 78% on OpenBind's triple success criteria. Closest next best is 54%.
2/ 2Å is too fuzzy. The number that matters: sub-1Å.
That's the real RMSD accuracy threshold that’s relevant to be useful in practice (for actually designing molecules and downstream predictions like potency).
→ Pearl: 60% → Best competitor: 27% → Most models: 1–13%
At this bar, Pearl beats every method — including classical docking that gets the bound crystal structure Pearl never sees.
3/ This target is hard.
A loop at the binding site shifts ~4Å when a ligand binds. The pocket doesn't even fit a ligand until the protein moves. Classical docking from the unbound structure can’t model this.
This also isn’t a target with similar structures in the PDB, so co-folding models can’t cheat. Unlike some benchmarks, this makes the OpenBind challenge a proper test of real-world utility.
Pearl predicts the motion from sequence alone and handles the new system with ease – including one compound placed to 0.28Å that no other zero-shot method solved.
4/ How we did it.
Synthetic data at training. Equivariant architecture. Novel inference time scaling. AI + physics scoring to converge the outputs.
This is what a foundation model that generalizes and works in actual drug discovery programs looks like — not one that memorizes the PDB.
Full write-up + figures 👇
This is overdue, and a small step. Our most recent experiments together with @AnthropicAI show that even with no access to any assay data (in fact publicly available capsid data is unhelpful in this case), the models are getting pretty good at predicting an actual biological dataset they've never seen. While packaging is the easiest trait to predict, it does empower downstream engineering tasks. We need not just passive control/awareness (without being alarmist), but highly proactive counter-measures.
Another closed-source antibody folding and design preprint showing very promising results, with successful design of VHHs against various targets as well as very accurate antibody-antigen complex prediction. No details on methods so lets hope it stays off biorxiv et al
Binder design has come of age thanks to generative models—but how can we access the wider array of dynamic, multistate protein functions, so elegantly employed by nature?
@mihirbafna14 and I are excited to share SwitchCraft, a framework for designing such functions. (1/7)
One of the most amazing things I’ve ever seen: a standing ovation for the full Daraxonrasib results
I feel inspired and energised, to put it mildly — we have a targeted therapy for pancreatic cancer now, and nothing is undruggable anymore
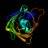
Eli Lilly has done it.
They've gone and made what seems to be a powerful, permanent gene therapy for LDL cholesterol.
That means they'll be able to effectively prevent most heart disease with a single infusion!
🚨 New blogpost
> ML Interatomic Potentials (MLIPs) are great for speeding up molecular modeling and bypassing expensive DFT-like ops.
> As they become mainstream, I see more work doing uncertainty quantification (UQ) for them.
> UQ for MLIPs is a lot more nuanced and doesn't quite translate as easily from UQ for traditional ML (vision, etc). Quite a few pitfalls to avoid.
I write about all this and more! Link in thread.
Next Tues (5/26) at 4PM ET, @ginaelnesr will present "Zero-shot design of a de novo metalloenzyme"
https://t.co/dv7A4dncG8
Sign up on our website for zoom links!
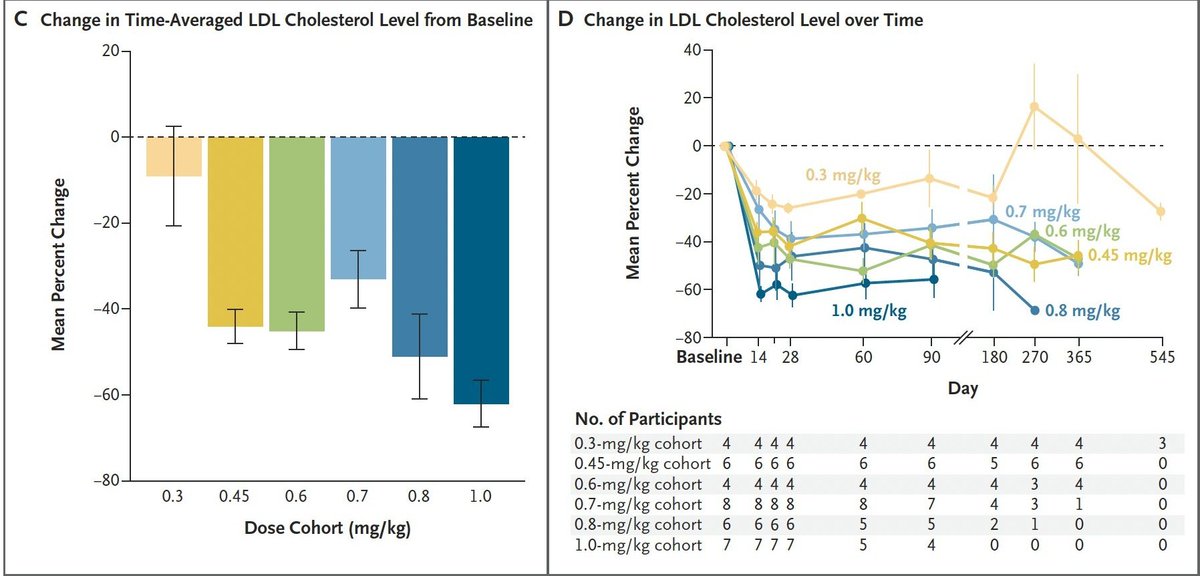
Excited to share our latest paper, out today @CellCellPress. We found that large pieces of the human genome can transfer between cells upon direct contact, endowing recipient cells with heritable phenotypic changes. (1/7)
https://t.co/SbshGhofN0
Introducing Carbon 🧬 a family of open generative DNA foundation models. Carbon-3B matches Evo2-7B while running 250x faster at inference. It can generate new DNA sequences and score the functional impact of mutations, zero-shot.
We borrowed a lot from how modern LLMs are trained, but DNA isn't language. Genomes are noisy, redundant, and shaped by evolution rather than communication. So we adjusted the recipe:
Tokenizer. Most genomic models tokenize at the nucleotide/character level, which blows up sequence length. BPE is the obvious LLM-style fix, but it doesn't behave well on DNA. We use deterministic 6-mer tokens (one token = 6 nucleotides): 6× shorter sequences and cheaper attention.
Training loss. With 6-mer tokens, cross-entropy scores a prediction that gets 5/6 nucleotides right the same as one that's completely wrong. This gets brittle late in training and produces loss spikes. We switch mid-training to a more flexible factorized loss (FNS).
Data. Genomes are mostly sparse, repetitive background. We curate down to a staged functional DNA + mRNA mixture, with every ratio chosen by ablation, like mixing a web corpus, but for biology.
We're releasing the models, training data, training code, evaluation suite, and a demo to play with.
More details in the technical report: https://t.co/RMzFmTAhhT
Demo to play with the model, with a biology primer for our ML friends ;) https://t.co/IcOQq7GKF4
I prompted the “deep literature” mode of Edison Scientific to write a comprehensive review on the biology of a gene that I know well.
Twenty five min later it sent a 17 page PDF. It was, as best as I can tell, flawless.
Folks, we are not in Kansas anymore.
Don’t at me - try it.
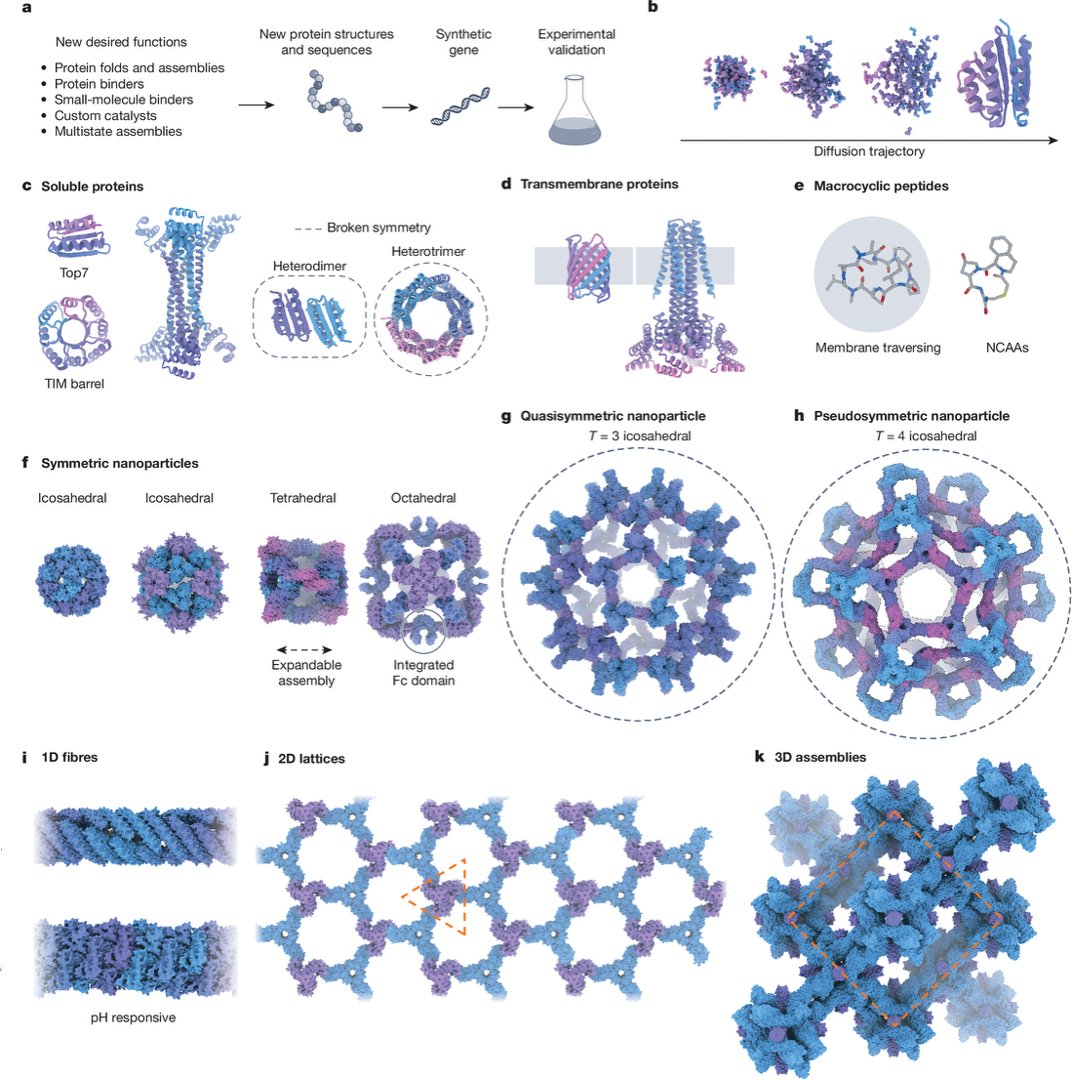
1/ Excited to share our new Review in @Nature:
“The past, present and future of de novo protein design.”
Here, we mainly focused on structure-guided protein design. The field is entering a new phase: now that we can design new proteins, what should we build next?
We benchmarked Arc Institute’s “MULTI-evolve” and found evidence that it learns a classical additive model, not epistasis.
Preprint: https://t.co/qYQxEXbJzM
Blog: https://t.co/BKCmRii3G1