Tutorial about raw data processing and quantitative analysis of CUT&Tag, CUT&RUN data available on https://t.co/ArFJBJl28j as well as GitHub https://t.co/LHzN8Xz386 Comments and suggestions are very welcomed! 😃
Samplot is a very useful tool for visually inspecting SVs in general and in rare disease contexts. Developed largely by @jon_belyeu and @ryanlayer's lab, it also includes a trained model that can automatically separate true and false variant calls with high accuracy.
To process 721,180,737 sequencing reads from a human/mouse single-cell RNA-seq experiment, Cell Ranger (python) will need ~2.5 gal of gas worth of energy to generate a count matrix while kallisto | bustools (c++) will need ~0.05 gal of gas (50x less). #speedmatters#methodsmatter
Very happy and proud to present: The GTEx papers. This set of 15 papers published today describes the final phase of this 10-year effort, providing the genomics community an atlas of genetic regulatory variants and a deep dive into the biology behind it. https://t.co/hntmcfwkoH
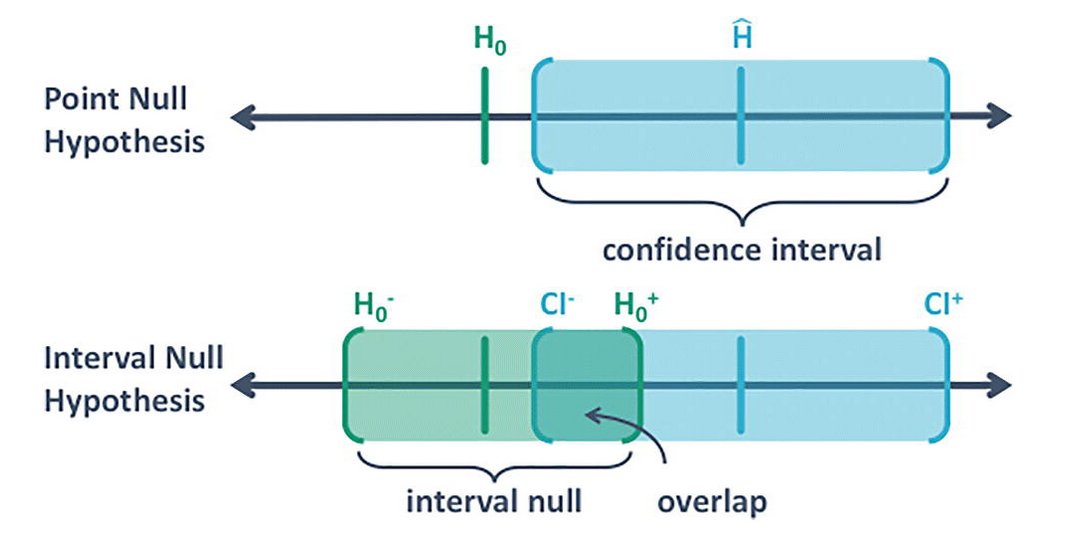
I just learned about second-generation p-values, by @StatEvidence.
They make conceptually more sense as they take into account that the null has an interval too. That there is a difference between null and practically null.
1/
h/t @f2harrell
https://t.co/W5pfWcWfn8
If you’re an academic you need a website so that people can easily find info about your research and publications. Here’s how to make your own website for free in around an hour using the blogdown package in #Rstats [UPDATED 2019 THREAD]
Wang, Sun, @fooliu, @XShirleyLiu and co present MAESTRO, for model-based analyses of transcriptome and regulome, for integrative analysis of single-cell RNA-seq and ATAC-seq data. It can automatically annotate cell type clusters, and infer regulators. https://t.co/SqY2jn83nA
Finally, our paper on integrative analysis of scRNA-seq and scATAC-seq is out (https://t.co/ck88jMbEsw)! Appreciate our community to give MAESTRO (https://t.co/U65DHmTHpR) a try and give us feedback. We will continue to maintain and improve accordingly.
my first preprint is up: Evaluating single-cell cluster stability using the Jaccard similarity index https://t.co/mjJoAMT1xl work with @timsackton@Bioinfomagics
@daweonline@coletrapnell and colleagues wrote a paper reporting empirical results on clustering by UMAP: https://t.co/Ir6pbdaHxu
I am not aware of theoretical results.
SRA plans to drop base quality. This would make resequencing data largely useless for var calling. There are better ways to save space: quality binning and CRAM. If you use reseq data from SRA, please voice yourself in this RFI (deadline in 2 weeks): https://t.co/Ii4d4wyH0s
Instead of providing static proteome snapshots, proximity labeling can now be used to study protein *translocation*. TurboID is expressed in the ER lumen of source tissue, then biotinylated proteins are harvested by dissection from destination tissue.
https://t.co/GjkEUdAt7S
A systematic map of over 50,000 protein-protein interactions is now available to better understand cancer and infectious diseases such as COVID-19 https://t.co/Go0BoMkhtV
gt = grammar of tables. It aims to do for tables what ggplot2 did for graphics. It’s still early days and tables are surprisingly complicated, but this is a very exciting package by a skilled developer! #rstats