@maxkreuzz@abhinadduri@yusufroohani@davey_burke@dhrvji So it's inevitable given current bio data. But since biological foundation models only need to capture biology (not general reasoning), we may not need comparable scale for it to be useful. Plus, we believe we've set up the right framework for scaling as more data comes. (2/2)
@maxkreuzz@abhinadduri@yusufroohani@davey_burke@dhrvji Thanks! You're right about the scale gap. All current human single-cell data is ~10¹⁰ tokens in stack, while modern LLMs train on >10¹³. Our model scales accordingly: 10⁸⁻⁹ params vs >10¹¹⁻¹² for SOTA LLMs—about 10⁶× smaller in total, hence the resource difference. (1/2)
Predicting cell state in previously unseen conditions such as disease or in response to a drug has typically required retraining for each new biological context. Today, Arc is releasing Stack, a foundation model that learns to simulate cell state under novel conditions directly at inference time, no fine-tuning required.
Super proud to present Stack — a foundation model that brings in-context learning to leverage and engineer cellular contexts, through innovations grounded in single-cell biology. Huge thanks to @yusufroohani@abhinadduri@dhrvji@davey_burke and Arc team! A great summary below:
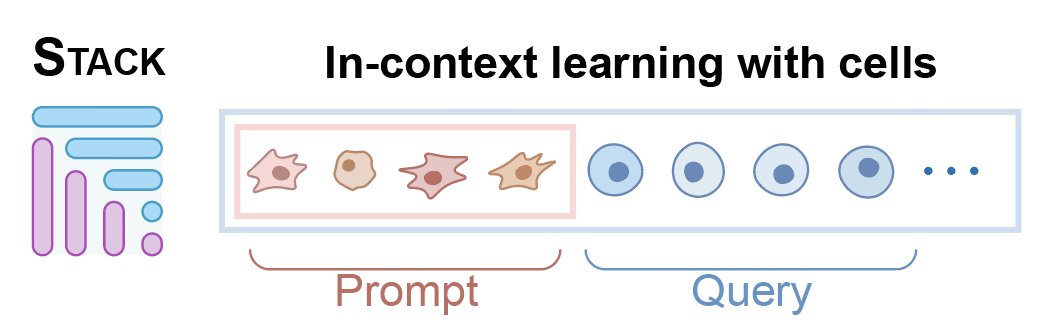
Why define conditions, donors or even *tasks* when we can just use cells themselves to guide model output
Presenting Stack, in-context learning using just cells!
Use cell context -> enhance its embedding
Engineer cell context ->modify its state
Led by the brilliant @Mingze7316
Open to DMs / chats about AI for science and academic job opportunities!
See my previous work on theoretically grounded single-cell and spatial omics AI models: https://t.co/NOkX8R23NX — with more to come.
I’ll be at #NeurIPS from Wed–Sun presenting our work https://t.co/DC8E7ZwSBf !
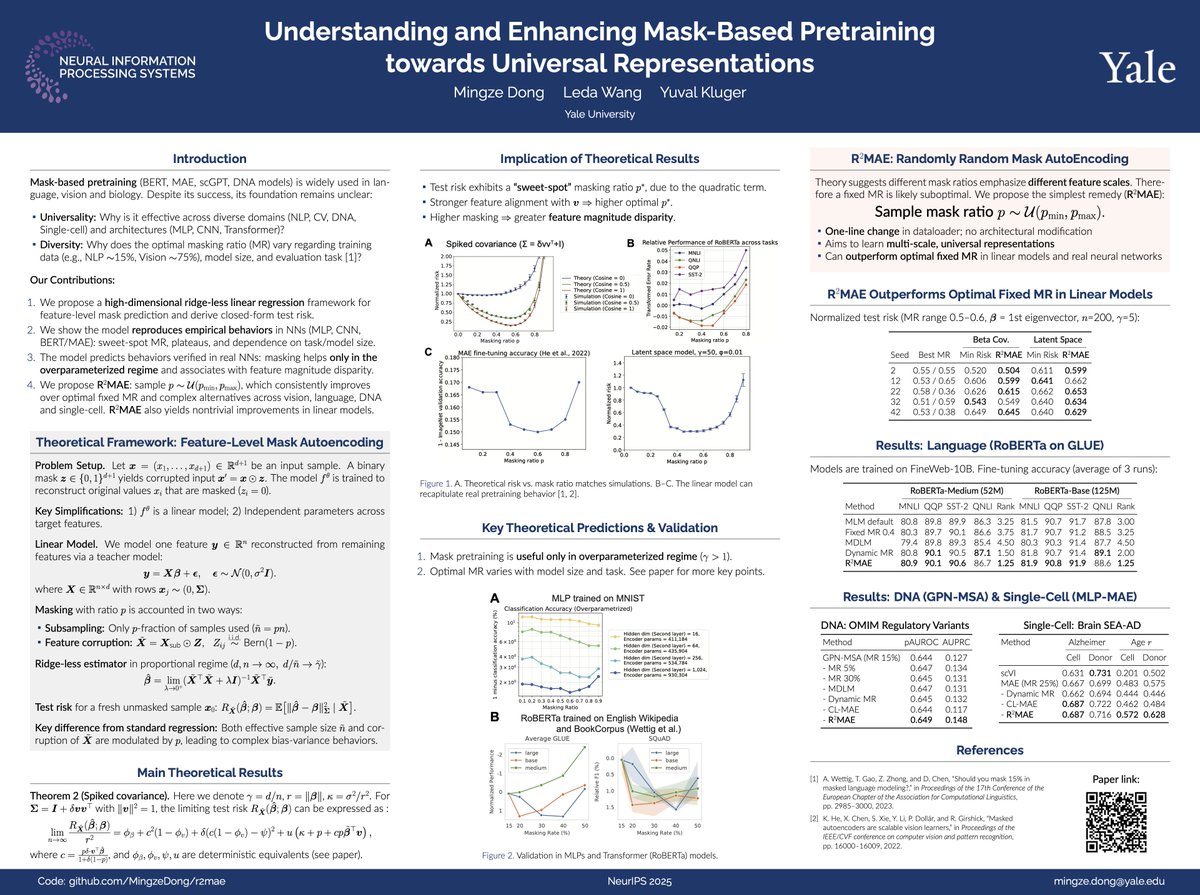
We build a high-dim linear model that explains all kinds of phenomena in mask-based pretraining, and from this framework propose R²MAE that improves pretraining across language, DNA, and single-cell.
Thrilled to share that our work is featured by @NatureComms as an editor's highlight in Computational and Theoretical Biology! Check the link below: https://t.co/z2y7IJpcp9
Out in @NatureComms! We tackle a core challenge in spatial omics—reliably disentangle spatial interactions from intrinsic cell properties, which requires identifiability. We built an identifiable deep learning framework SIMVI (with proofs!) to solve this: https://t.co/JnJnDRQnIF
By identifiability, SIMVI uniquely enables inference of “spatial effects” at a single-cell level, empowering biological discoveries. Please refer to our manuscript (and the 44-page SI) for more details and applications. Many thanks for the support @YaleCBB@RongFan8@Klugerlab!
@Ella_Maru Thanks for the question! Short answer: Yes. If the lineage is space-independent, intrinsic variation would capture and disentangle it from niche; if space-dependent, our relevant case study (Fig. 5) shows SIMVI can reveal spatial-dependent states and differentiate from niches.
Many thanks to all co-authors whose contributions make this work possible! Please check our manuscript for details and more results: https://t.co/Y0sUV0ewoN
N/N.
Thrilled to share our preprint: https://t.co/Y0sUV0f4el. Long story short: we found a way (scShift) leveraging massive single-cell atlases to build powerful zero-shot biological state extractors. Its performance scales with dataset diversity after an “emergence threshold”. 1/N
Summary: scShift demonstrates 4 important properties for next-generation single-cell models: 1) zero-shot, 2) disentanglement, 3) scaling, and 4) unsupervised. It facilitates analyses of biological states at all levels. The novel idea may lead to various future extensions. 11/N