In conclusion:
🐮 @nanopore sequencing as rapid new tool to determine acquired AMR & support evaluation of ECOFF values
🐮 current approach allows shortening of the present sample-to-result workflow (IDx & AST)
🐮 future goal = immediate IDx & AST without pre-enrichment
6/6 🧵
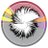
🟩🟦🟩 Genotypes allowed for re-evaluation of various ECOFF methods 📈
🧐 Existence of single gene mutants (ENRO; n=3) within the Wild-Type population, are overlooked during phenotypic testing. However, the genotype 🧬 provides a higher resolution.
#Antibiotics#wiltype 5/6
No quick Antimicrobial Susceptibility Tests are available for M. bovis, so quick & accurate sequencing-based approaches are important to
- provide appropriate & rapid antimicrobial treatment in the framework of AMR
- prevent disease from spreading and/or becoming chronic
4/6
A k-mer-based GWAS analysis allowed to link M. bovis genotypes 🧬 with phenotypes 🧫 based on different ECOFF thresholds. This highlighted potential genetic markers for:
1- macrolides (GAM, TYLT & TIL)
2- fluoroquinolone (ENRO)
3- tetracyclines (OXY & DOXY)
3/6
📇 The manuscript can be found here:
https://t.co/aQKJ9lFT5l
🛠 100 @nanopore High-Quality & Complete bacterial genomes were generated using an optimized M. bovis 🐮 specific Bonito model (Vereecke et al., 2020 BMC Bioinformatics) 📄 2/6
#Bonito@iiSeymour#genomics@kehoste
A new week, so a new co-first shared publication from the @pathosense lab 🥳 in @JournalSpectrum
"Genome-Wide Association Study Reveals Genetic Markers for Antimicrobial Resistance in Mycoplasma bovis" 🐮
⏭Read further to learn more in this 🧵 1/6
#sequencing#mycoplasma
"Evaluation of @nanopore sequencing as a diagnostic tool for the rapid identification of Mycoplasma bovis from individual and pooled respiratory tract samples"
⏩ our latest publication in @JClinMicro
⏩ A nice collaboration between @UGent_VetMed & @pathosense
🔽Check the 🧵
New weekend, new #seaofgreen on our MinION. 18h into sequencing and a 10.5 Gbp output so far (still 30h ahead and 83% of @nanopore pores still sequencing).
👉 RT Bacterial IDx ✅
🐷 RT Bacterial (Virulence) Typing ✅
⭕️ RT Bacterial AMR profiling ✅
🧬 Complete HQC genomes ✅
Ready for this #nanoporeconf 🧬 today & tomorrow! I am very excited to hear about new @nanopore stuff that will be released, but also new applications & research from experts in the field!
Oh, and don't forget to check my Mini Theatre talk 👇 on Mycoplasma bovis 🐄
@pathosense
Finally out! https://t.co/MPzjPvhgPX
See how we have used long-read metagenomics with Nanopore sequencing to profile a canine fecal microbiome sample and retrieve eight single-contig HQ MAGs 🐕💩🦠🧬
@prd_science
@Quinovines
@NormaFabregas@olga_francino
Well hello there 💙💚 Let’s finish this Friday with this bacterial 🧬 sequencing run using the @nanopore Rapid Barcoding Kit! Never thought we would reach these N50 values (> 27kbp) & this caption is only after 30 min sequencing! 🤪🥳
@theuns_bas#AMR#Diagnostics
Check out this talk preview from @methenickname. At #nanoporeconf, he'll demonstrate how nanopore sequencing was used to identify resistance markers & speed up antimicrobial susceptibility analysis in veterinary samples 🐄.
Register to hear in full: https://t.co/smeqHENW0D
Mycoplasma bovis causes severe respiratory issues & poses a mortality risk to cattle. At #nanoporeconf, @methenickname will demonstrate how nanopore sequencing was used to identify resistance markers & speed up antimicrobial susceptibility analysis.
https://t.co/KTybzeZ67Z
🦠Fresh bacterial release in @Micro_MDPI
Together with @theuns_bas, @pathosense & @UGent_VetMed we showed the presence of BSBL-producing Enterobacteriaceae in 💩 of Zoo mammals with @nanopore sequencing. A potential zoonotic & public health threat? 1/4
https://t.co/RJPxKVn3fl
#OutbreakAlert 🐎 February-March: Europe's biggest EHV-1 outbreak was reported in Spain (17 dead & >80 infected 🐎). The #Nauwynck lab isolated EHV-1 from 🇧🇪 suspected 🐎 & by the end of the week @pathosense made complete (150kbp) genomes available with @nanopore sequencing 1/6
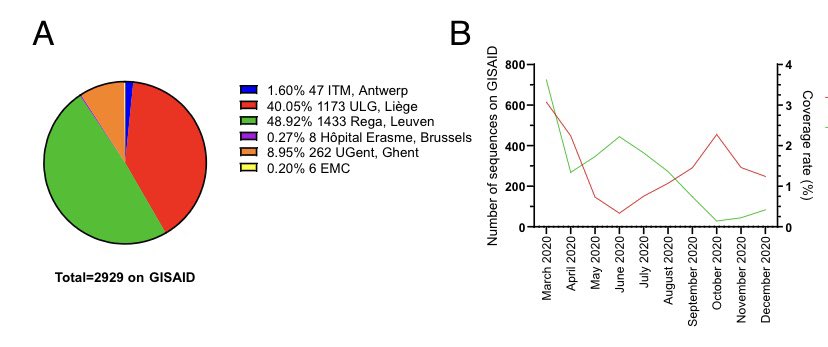
A quick analysis for Belgium to show how we are doing, and what should be improved. This is all based on public data available on GISAID In panel A you see 98% of the genomes were done by either REGA (@MaesPiet ), Liège (@keithdurkin) and Ghent (@theuns_bas).