😎Binder #5 from PallAtom v2 was designed using a head-and-tail binding strategy that simultaneously optimizes for target-binding affinity and E3 inhibitory potency. We look forward to conducting additional E3 activity inhibition assays to further validate its functional efficacy
RBX1 Binder Design Competition results are out!
Against BindCraft's dominant performance, PallAtom stands out as the strongest Diffusion-based Model.
🎉Congratulations to my old crew!
😎Binder #5 from PallAtom v2 was designed using a head-and-tail binding strategy that simultaneously optimizes for target-binding affinity and E3 inhibitory potency. We look forward to conducting additional E3 activity inhibition assays to further validate its functional efficacy
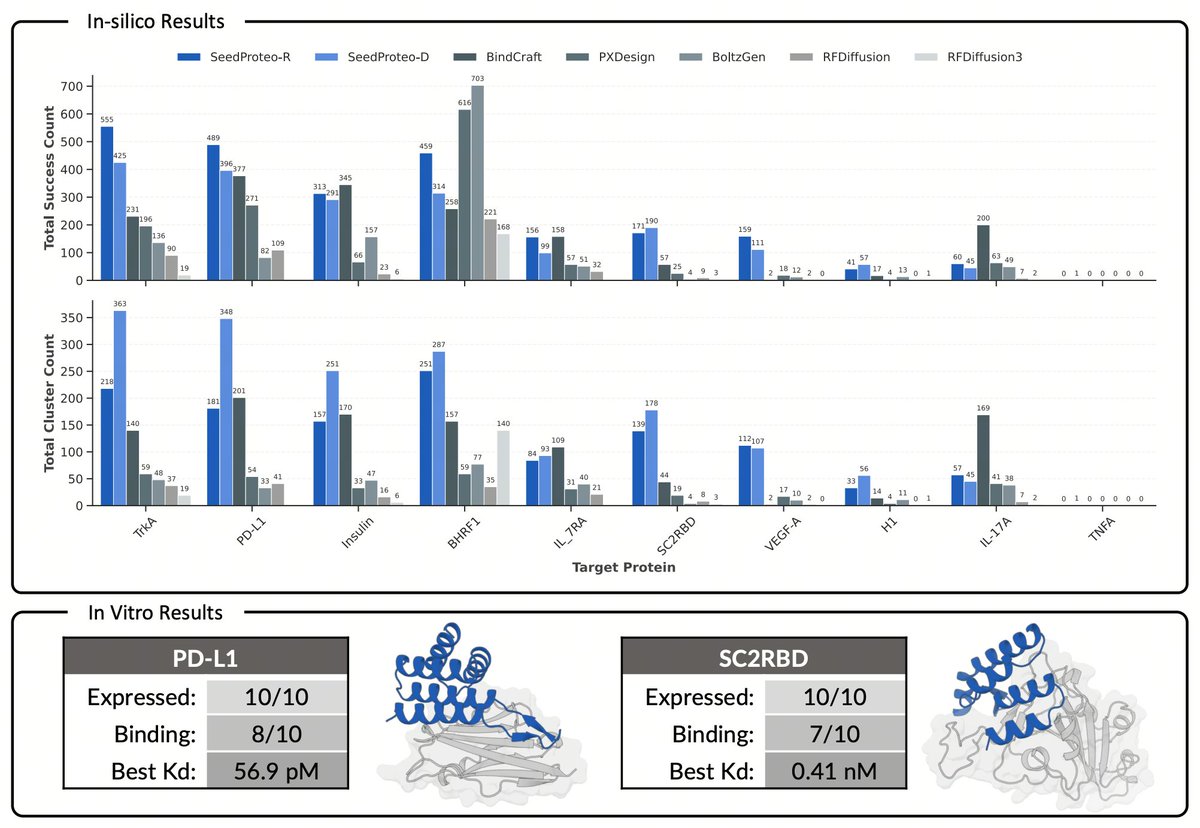
Here is SeedProteo, our latest diffusion-based model for de novo all-atom protein design from ByteDance Seed! Our server is now live — feel free to give it a try! https://t.co/Tqsu2VPmWw
Introducing PAR: We’ve been exploring autoregressive protein generation lately — generating protein structures in an autoregressive language modeling style is surprisingly fun.
🧬 Protein Autoregressive Modeling (PAR), ByteDance Seed
We introduce PAR, the first multi-scale AR framework for protein backbone generation.
Autoregressive modeling of 3D protein structures has long been considered difficult.
Here’s how we make it possible with PAR. 👇
https://t.co/VYN6PJim2p
Excited to share STAR-MD: a scalable autoregressive diffusion model that generates stable, high-quality protein MD trajectories at microsecond timescales, where existing methods fail catastrophically.
Accepted at ICLR 2026! Links and details in the thread 🧵👇
1/7
Introducing SeedFold from ByteDance Seed, a biomolecular structure prediction model that outperforms AlphaFold3 on protein-related folding tasks. https://t.co/4a93w6VLJL
We’re thrilled to announce our collaboration with @EliLillyandCo.
We’ll be deploying Chai’s AI models to power biologics discovery across a broad set of drug programs at Lilly.
This is a major step forward in pharma’s adoption of AI-native R&D.
Announcing a new @GPU_MODE kernel writing competition: our first featuring both NVIDIA and AMD hardware!
The first problem will be the Triangle Multiplication operator essential to the AlphaFold 🧬 models! It's a particularly tricky problem with no good public implementation!
Benchmarking Sequence-Based and AlphaFold-Based Methods for pMHC-II Binding Core Prediction: Distinct Strengths and Consensus Approaches
1. This study benchmarks four recent methods for pMHC-II binding core prediction: two sequence-based (NetMHCIIpan, DeepMHCII) and two AlphaFold-based (AF2-FT, AF3). AF3 achieved the highest positive recall (0.86), while NetMHCIIpan excelled in predicting non-binders, with the highest negative recall (0.93).
2. The study demonstrates that AlphaFold-based methods are excellent at predicting positive binders but tend to misclassify non-binding peptides. NetMHCIIpan, in contrast, is highly precise in identifying non-binders but struggles with positive recall, missing true binders.
3. A consensus approach combining AlphaFold-based methods’ strengths for positive prediction and NetMHCIIpan’s non-binder filtering improves overall accuracy and precision. This consensus approach achieves an F1 score of 0.93 with perfect recall, highlighting the potential for improved pMHC-II binding core predictions.
4. The study also identifies challenges with longer peptides and chimeric MHCs, both of which reduce prediction accuracy, especially for sequence-based methods.
5. Future improvements should focus on enhancing AlphaFold-based models’ ability to filter out non-binders and refining NetMHCIIpan’s binder prediction capabilities to strike a better balance between precision and recall.
📜Paper: https://t.co/4g326P8uSw
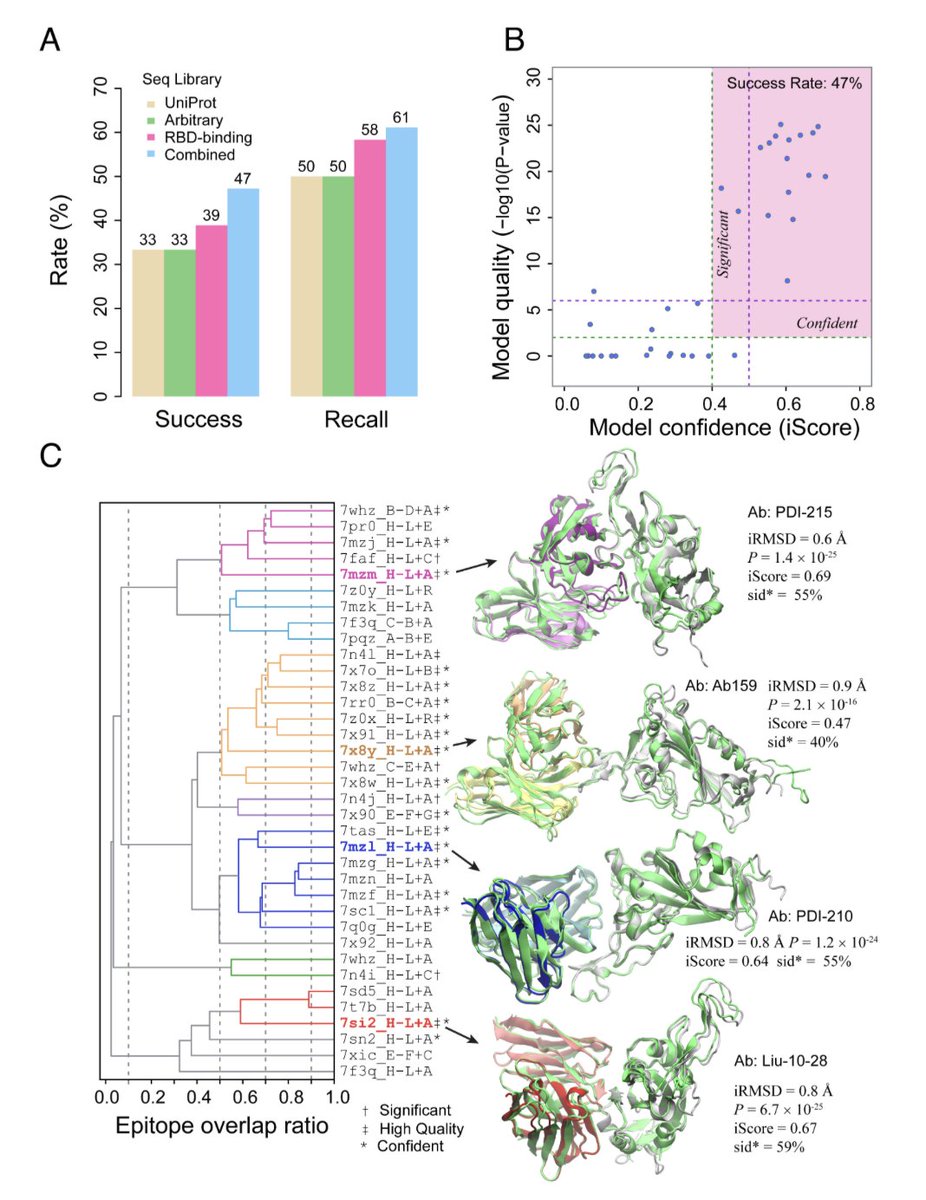
Improved deep learning prediction of antigen–antibody interactions @PNASNews
1/ This paper presents a breakthrough in predicting antigen–antibody interactions using deep learning models, particularly focusing on the spike protein of SARS-CoV-2. It evaluates the AF2Complex model's accuracy in predicting the structure of antibody-antigen complexes.
2/ One key innovation is the use of B cell sequencing data to improve prediction accuracy. By employing known RBD-binding antibodies, the AF2Complex model significantly outperforms previous approaches, achieving a top prediction accuracy of 61% on a benchmark of 36 antibodies.
3/ The study demonstrates that the deep learning approach can distinguish true antigen-binding antibodies from randomly selected antibodies with remarkable precision. For example, the model identifies true positives with a precision of 90% while maintaining a recall of 25%.
4/ This deep learning method offers a robust tool for predicting antibody-antigen interactions without requiring experimental structural data, making it highly valuable for vaccine design and therapeutic antibody development, especially in cases like COVID-19.
5/ The study highlights the AF2Complex model's capability to detect antibodies that target diverse epitopes, proving its utility for complex immune-related tasks and expanding the potential applications of deep learning in immunology and drug development.
💻Code: https://t.co/B96Mf2j4NK
📜Paper: https://t.co/VHMkk5tDyC
1/X Pallatom is an end-to-end all-atom protein generation model that directly learns the distribution of atomic coordinates. We believe optimizing the P(all-atom) may open a new way for de novo protein design.
code(ing):https://t.co/QqNhRKmpGN
bioxiv:https://t.co/2I9Tv6DPra