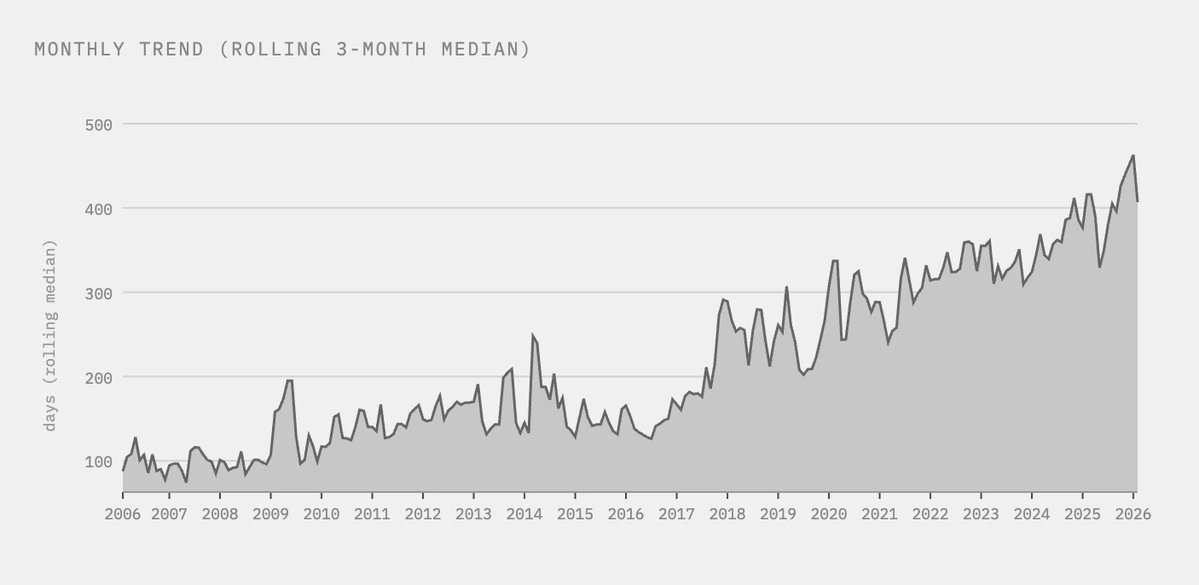
No scaling laws for single-cell foundation models: when bigger atlases stop teaching the model anything
In language and vision, the recipe has been simple: more data, bigger models, better performance. Single-cell biology borrowed that playbook. Foundation models for transcriptomics jumped from 1 million cells to atlases of over 100 million, on the assumption that scale would unlock the same gains. Alan DenAdel and coauthors put that assumption to the test, and the result is sobering.
Working from a 22.2-million-cell corpus, they pretrained 400 models across five architectures (from PCA and a variational autoencoder up to the Geneformer transformer) and ran 6,400 evaluation experiments. They varied not just dataset size (1% to 75%) but also diversity, using cell-type re-weighting and geometric sketching to deliberately enrich rare cell types and transcriptional states.
The finding: performance saturates almost immediately. On cell-type classification, batch integration, and perturbation prediction, most models hit their ceiling at roughly 1% of the corpus, about 200,000 cells. Beyond that, adding millions more cells changed essentially nothing. More diversity didn't help. Even spiking in genome-scale Perturb-seq data, to give the models perturbed phenotypes rather than just healthy ones, failed to move the needle. Larger models did score better overall, but they too plateaued early on data.
Two points stood out. Simple baselines (PCA, logistic regression) often matched or beat the transformers. And the strongest model, SCimilarity, won not because of size but because its contrastive training objective is aligned with the downstream task. For single-cell data, what you train on and how you frame the objective matters far more than how much you collect.
This reframes a quiet but expensive habit. In drug discovery, biotech, and any pipeline leaning on cell atlases, the instinct to keep scaling pretraining corpora may be burning compute for no return. The real leverage sits elsewhere: curating high-quality, task-relevant data and matching the training objective to the actual question you're trying to answer.
Paper: DenAdel et al., journal license | https://t.co/X7GxoxF5U5
This new paper is probably the most prominent example to date of how linking genetic variation to cell-level, rather than tissue-level, gene expression can transform the interpretation of GWAS signals.
The study generates a single-cell eQTL resource from intestinal biopsies and blood samples from 421 individuals, including 125 with inflammatory bowel disease (IBD)👇
Last week we've gathered 80 colleagues from the biotech industry to discuss how they're integrating AI agents into data teams.
We wrote seven quick takeaways on how agents are already transforming biotech >>
Excited to share Decima, out now in @naturemethods! 🎉
Existing seq-to-function models predict bulk expression. Decima goes further: it predicts gene expression in specific cell types and disease states from DNA sequence alone — trained on 22M+ single cells.
Applications: cis-regulatory mechanisms, cell-type-resolved variant effect prediction, and designing context-specific regulatory DNA
Analyzing phenotypes on the wrong scale reduces power and creates spurious genetic interactions. SIQReg, a new method from my postdoc Zhenhong Huang, learns an optimal phenotype scale to fix this: https://t.co/u8QzlAEbNv
New preprint on a controversial topic:
Through which socioeconomic mechanisms do individuals with higher genetic predisposition for educational attainment end up with higher income over their working life?
We study this using uniquely rich Finnish data.
New preprint led by Hrushikesh Loya, Leo Speidel, and I where we introduce GhostBuster! https://t.co/MgfPEVeHQJ
Our method uses genealogies to find "ghost" ancestries hidden within DNA. We find both modern humans, Neanderthals formed as mixtures of two ancient hominin groups
My full interview on TBPN where I talk about:
-polygenic prediction in IVF
-regulation of genetic testing in IVF
-the utility of biobanks
-my own experience as a cancer patient
-the future of reproductive medicine
A much better discussion than Kian Sadeghi on Tucker...
I usually dump on EVO2 quite a bit (mostly since it's been overhyped to death beyond it's actual capabilities), but this is nice work showing that a better embedding approach with supervised probing can deliver strong performance on coding variant effect prediction. 1/
Our Human Multiomic Development Atlas paper is out in Nature today! A heart-felt "thank you" to all co-authors for their tireless work on this complex yet exciting project! Congrats all! https://t.co/iUiZz00KOt
Delighted to share our latest research from the @23andMeResearch Team, just published in @Nature
! We looked at data from >27,000 participants to uncover how human genetics influences weight loss efficacy and side effects of GLP-1 medications like semaglutide. A thread 🧵👇
StratGWAS is our new tool for more efficient GWAS of heterogeneous diseases. Instead of treating cases equal, it weights them based on relevant phenotypic information such as medication use, age of onset or recruitment strategy. Full details on MedrXIv https://t.co/J6ANjdmztL.
Discover PARM, a deep-learning model predicting human promoter activity with precision. Major leap for genomics, decoding cellular responses. PMID:41639451, Nature 2026, @Nature https://t.co/CB3gO9QnZm #AI#Pharma#BioMed#RNA#ASHG#ESHG
Just posted a preprint on a huge new single-cell study of Parkinson’s and its application to understanding noncoding variation. 🧵below. Led by Shreya Menon and Adam Turner and in collab w/ GP2 (@ASAP_Research), @BelloyMichael, @ZihHuaFang and others. https://t.co/tBKZgq5bQ7
Our new preprint “Learning lifetime disease liability reveals and removes genetic confounding in electronic health records” is now online! Link to paper: https://t.co/7UNDZkHvP4 This work is led by my postdoc @diyazheng_ and it’s our first project at @ETH_BSSE :)
Thread 1/n
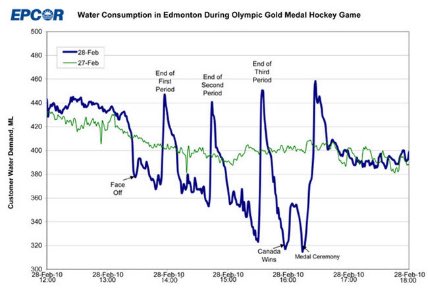
15 years ago the entire nation of Canada synced up there piss breaks during the gold medal game. This is what were up against folks we have to lock in Sunday
We're hiring a postdoc in my group at @calico! We develop deep learning methods for regulatory genomics to predict how every nucleotide in the genome affects cell-type-specific gene regulation.
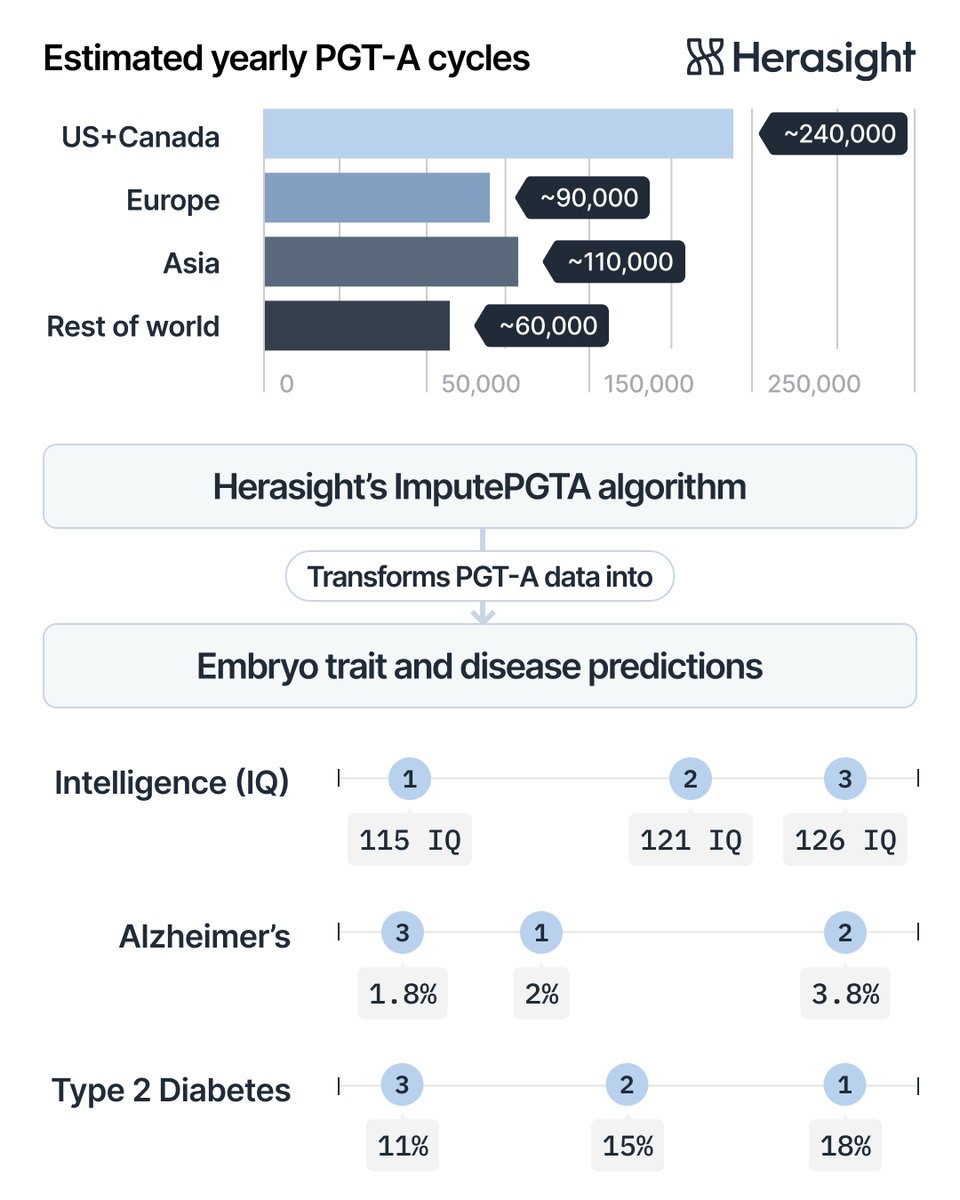
Today we’re announcing an algorithmic breakthrough.
Herasight’s ImputePGTA algorithm has enabled couples around the world to access polygenic embryo testing from routine IVF data (PGT-A).
Now it yields substantially higher accuracy, especially for underrepresented ancestries